
A Strainer-Based Platform for the Collection and Immunolabeling of Mouse Intestinal Organoids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
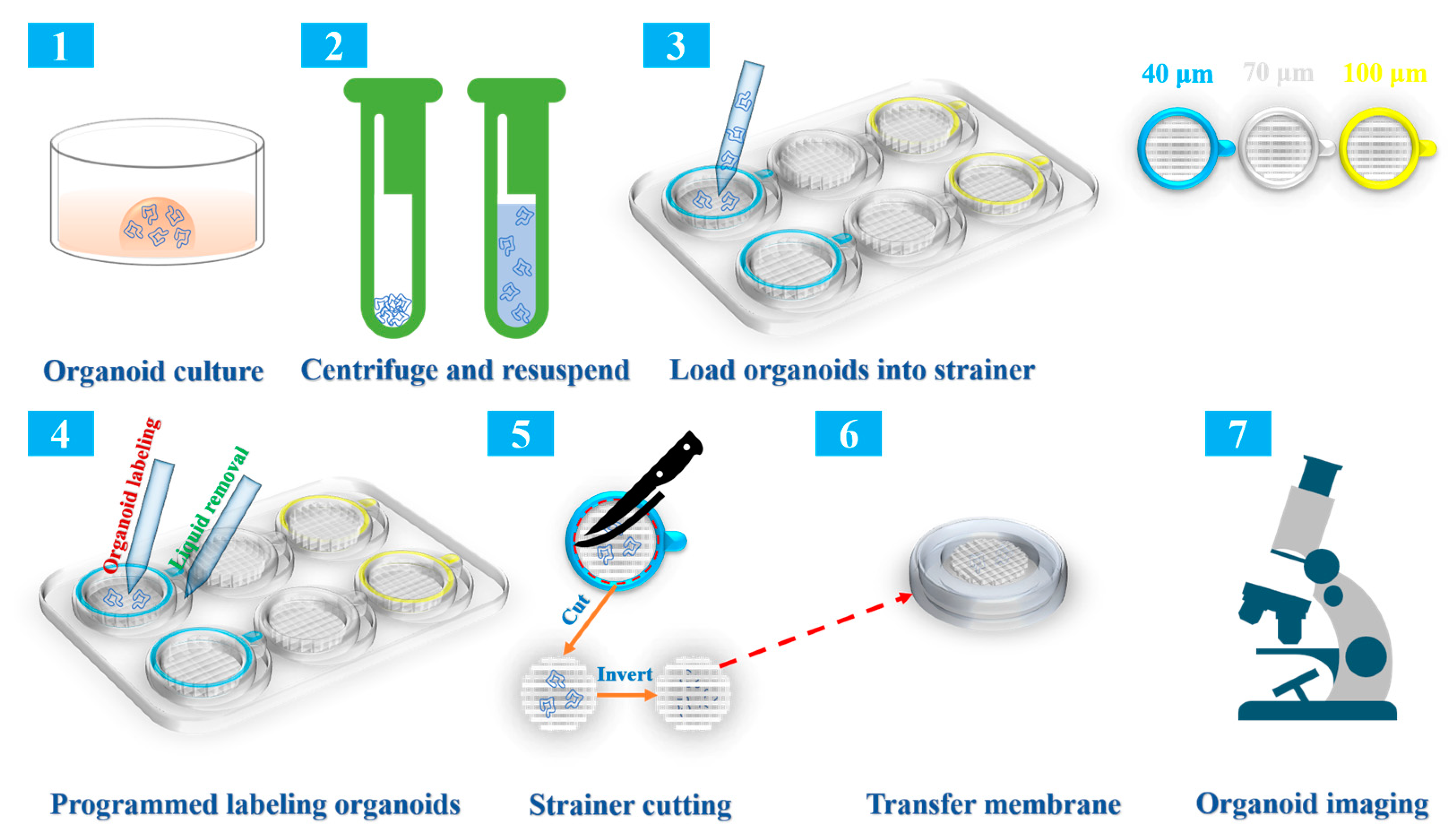
2.1. Overview of the Collection, Immunolabeling, and Three-Dimensional (3D) Imaging Protocols
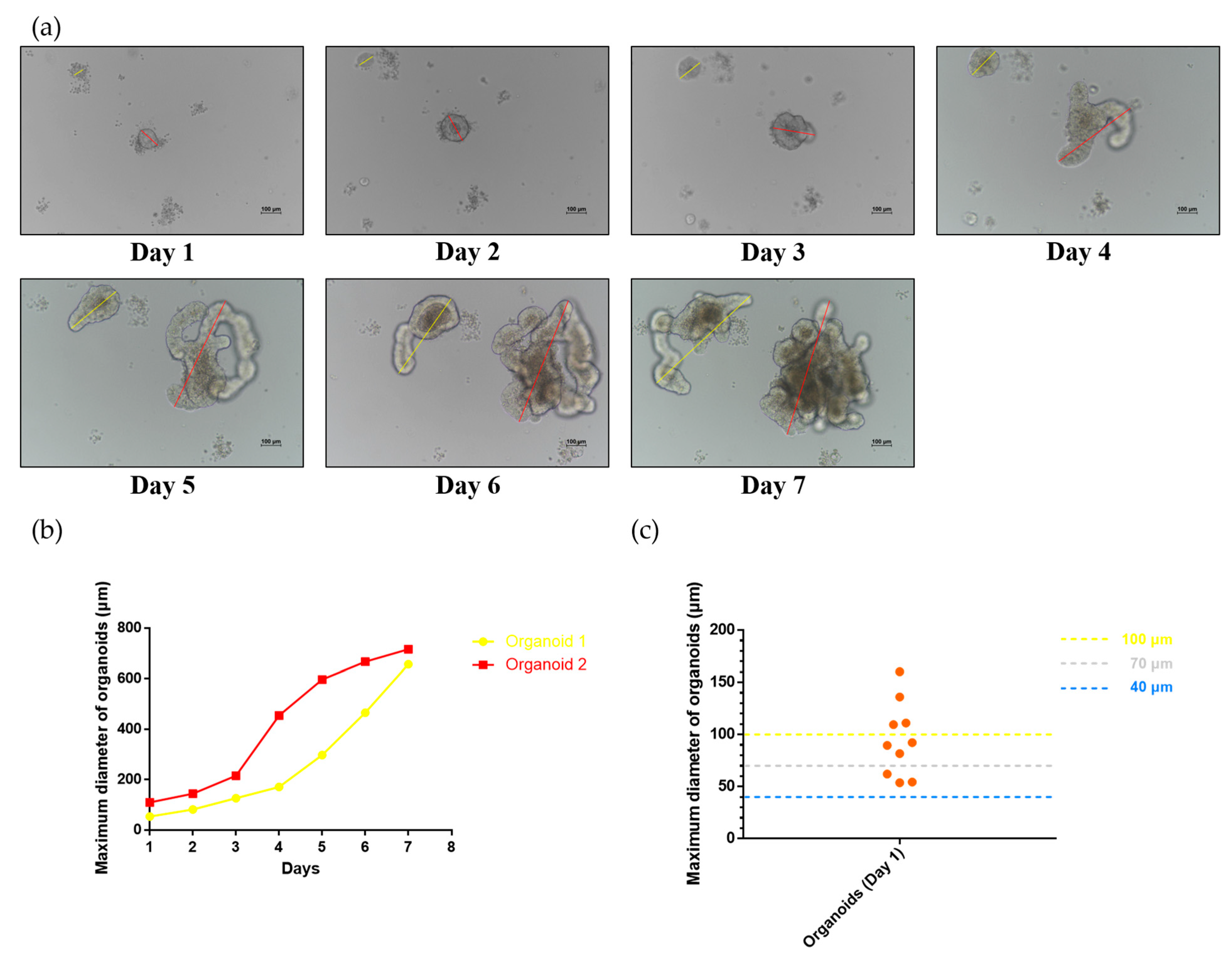
2.2. Development of Small Intestinal Organoids in the Mouse
2.3. Construction of a Strainer Platform and Programmed Labeling of Organoids
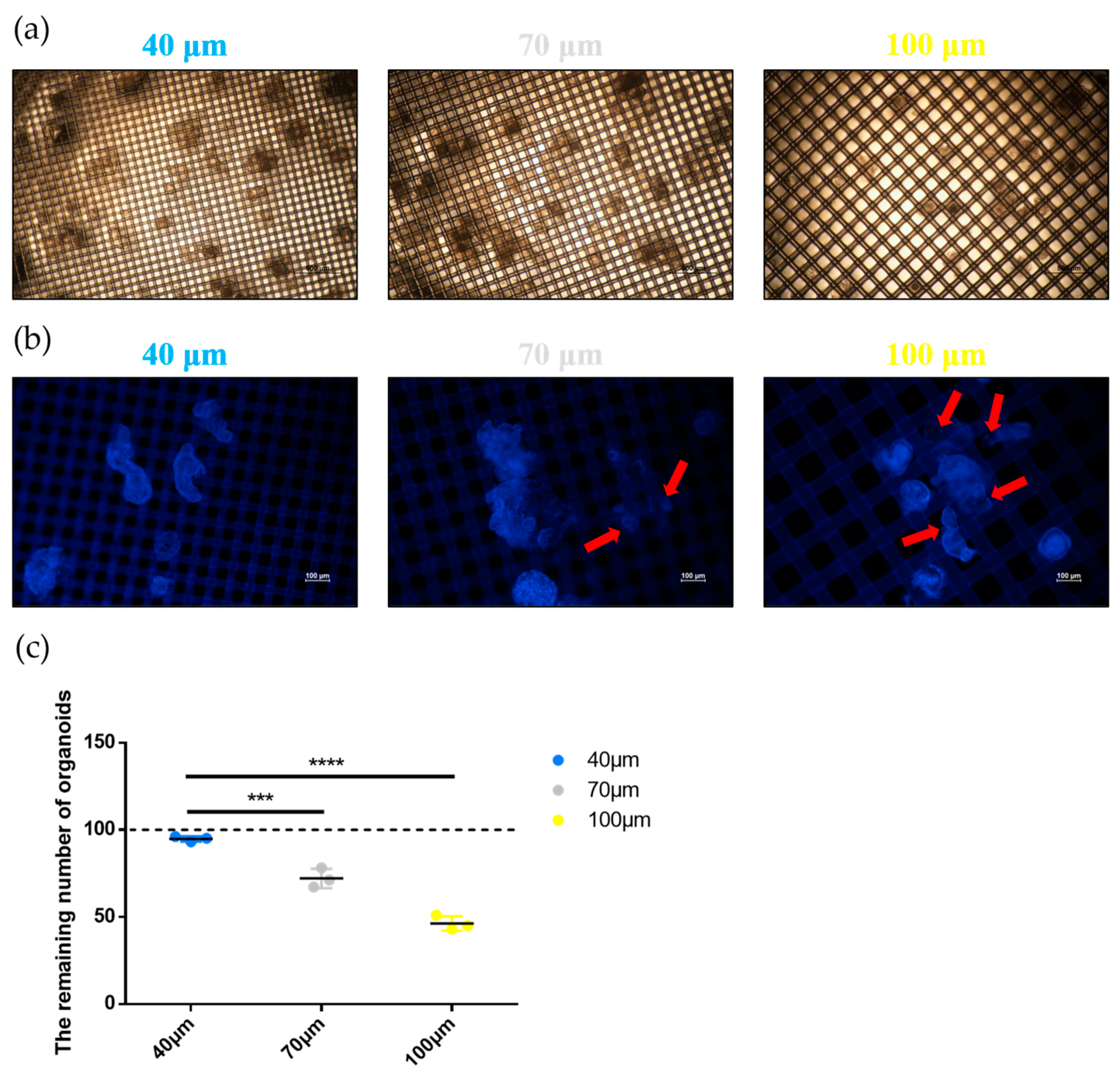
2.4. Evaluation of the Strainer Platform
2.5. 3D Imaging of Small Intestine Organoids
3. Discussion
4. Materials and Methods
4.1. Organoid Harvest and Culture
4.2. Organoid Passage and Collection
4.3. Strainer Loading of Organoids
4.4. Programmed Labeling of Organoids
4.5. Processing and Imaging of Organoids
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poirier, E.Z.; Vignuzzi, M. Virus population dynamics during infection. Curr. Opin. Virol. 2017, 23, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Tan, Z.W.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts in tumor microenvironment—Accomplices in tumor malignancy. Cell Immunol. 2019, 343, 103729. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Gao, Z.; Zhang, Q.; Gu, C. The mechanism of berberine alleviating metabolic disorder based on gut microbiome. Front. Cell Infect. Microbiol. 2022, 12, 854885. [Google Scholar] [CrossRef]
- Chen, Z.; Li, G.; Liu, J. Autonomic dysfunction in Parkinson’s disease: Implications for pathophysiology, diagnosis, and treatment. Neurobiol. Dis. 2020, 134, 104700. [Google Scholar] [CrossRef]
- Ma, G.L.; Qiao, Z.L.; He, D.; Wang, J.; Kong, Y.Y.; Xin, X.Y.; Wen, F.Q.; Bao, S.J.; Ma, Z.R.; Wang, F.S.; et al. Establishment of a low-tumorigenic MDCK cell line and study of differential molecular networks. Biologicals 2020, 68, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.P.; Su, T.C.; Yang, M.J.; Chen, W.T.; Siao, A.C.; Huang, L.R.; Lin, Y.Y.; Kuo, Y.C.; Chung, J.F.; Cheng, C.F.; et al. Green tea epigallocatechin gallate suppresses 3T3-L1 cell growth via microRNA-143/MAPK7 pathways. Exp. Biol. Med. 2022, 247, 1670–1679. [Google Scholar] [CrossRef]
- Robinson, N.B.; Krieger, K.; Khan, F.M.; Huffman, W.; Chang, M.; Naik, A.; Yongle, R.; Hameed, I.; Krieger, K.; Girardi, L.N.; et al. The current state of animal models in research: A review. Int. J. Surg. 2019, 72, 9–13. [Google Scholar] [CrossRef]
- Moro, C.A.; Hanna-Rose, W. Animal Model Contributions to Congenital Metabolic Disease. Adv. Exp. Med. Biol. 2020, 1236, 225–244. [Google Scholar]
- Fricker, M.; Deane, A.; Hansbro, P.M. Animal models of chronic obstructive pulmonary disease. Expert Opin. Drug Discov. 2014, 9, 629–645. [Google Scholar] [CrossRef]
- Goyal, S.N.; Reddy, N.M.; Patil, K.R.; Nakhate, K.T.; Ojha, S.; Patil, C.R.; Agrawal, Y.O. Challenges and issues with streptozotocin-induced diabetes—A clinically relevant animal model to understand the diabetes pathogenesis and evaluate therapeutics. Chem. Biol. Interact. 2016, 244, 49–63. [Google Scholar] [CrossRef]
- Zak, O.; O’Reilly, T. Animal infection models and ethics—The perfect infection model. J. Antimicrob. Chemother. 1993, 31 (Suppl. D), 193–205. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Liu, Y.; Yang, F.; Chen, G.; Fang, Y.; He, X.; Lou, Z.; Jia, H.; Jing, Z.; Li, W. Emerging evidence for poxvirus-mediated unfolded protein response: Lumpy skin disease virus maintains self-replication by activating PERK and IRE1 signaling. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2023, 37, e22902. [Google Scholar] [CrossRef] [PubMed]
- Almeqdadi, M.; Mana, M.D.; Roper, J.; Yilmaz, O.H. Gut organoids: Mini-tissues in culture to study intestinal physiology and disease. Am. J. Physiol. Cell Physiol. 2019, 317, C405–C419. [Google Scholar] [CrossRef] [PubMed]
- Idris, M.; Alves, M.M.; Hofstra, R.M.W.; Mahe, M.M.; Melotte, V. Intestinal multicellular organoids to study colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188586. [Google Scholar] [CrossRef] [PubMed]
- Norkin, M.; Huelsken, J. TORNADO-seq: A Protocol for High-Throughput Targeted RNA-seq-Based Drug Screening in Organoids. Methods Mol. Biol. 2023, 2650, 65–75. [Google Scholar] [PubMed]
- Malheiro, A.; Harichandan, A.; Bernardi, J.; Seijas-Gamardo, A.; Konings, G.F.; Volders, P.G.A.; Romano, A.; Mota, C.; Wieringa, P.; Moroni, L. 3D culture platform of human iPSCs-derived nociceptors for peripheral nerve modeling and tissue innervation. Biofabrication 2021, 14, 014105. [Google Scholar] [CrossRef]
- Cho, J.; Lee, H.; Rah, W.; Chang, H.J.; Yoon, Y.S. From engineered heart tissue to cardiac organoid. Theranostics 2022, 12, 2758–2772. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.Y.; Ju, X.C.; Li, Y.; Zeng, P.M.; Wu, J.; Zhou, Y.Y.; Shen, L.B.; Dong, J.; Chen, Y.J.; Luo, Z.G. Generation of vascularized brain organoids to study neurovascular interactions. Elife 2022, 11, e76707. [Google Scholar] [CrossRef]
- Staab, J.F.; Lemme-Dumit, J.M.; Latanich, R.; Pasetti, M.F.; Zachos, N.C. Co-Culture System of Human Enteroids/Colonoids with Innate Immune Cells. Curr. Protoc. Immunol. 2020, 131, e113. [Google Scholar] [CrossRef]
- Bergmann, S.; Lawler, S.E.; Qu, Y.; Fadzen, C.M.; Wolfe, J.M.; Regan, M.S.; Pentelute, B.L.; Agar, N.Y.R.; Cho, C.F. Blood-brain-barrier organoids for investigating the permeability of CNS therapeutics. Nat. Protoc. 2018, 13, 2827–2843. [Google Scholar] [CrossRef]
- Lukonin, I.; Zinner, M.; Liberali, P. Organoids in image-based phenotypic chemical screens. Exp. Mol. Med. 2021, 53, 1495–1502. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Taguchi, M.; Sumi, S.; Oka, K.; Okamura, K. Establishment of a novel protocol for formalin-fixed paraffin-embedded organoids and spheroids. Biol. Open 2023, 12, bio059882. [Google Scholar] [CrossRef]
- Lee, A.H.; Elliott, D.M. Freezing does not alter multiscale tendon mechanics and damage mechanisms in tension. Ann. N. Y. Acad. Sci. 2017, 1409, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Alieva, M.; Wellens, L.M.; Ariese, H.C.R.; Jamieson, P.R.; Vonk, A.M.; Amatngalim, G.D.; Hu, H.; Oost, K.C.; Snippert, H.J.G.; et al. High-resolution 3D imaging of fixed and cleared organoids. Nat. Protoc. 2019, 14, 1756–1771. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.J.; Dellavance, A.; Baldo, D.C.; Rodrigues, S.H.; Grecco, M.; Prado, M.S.; Agustinelli, R.; Andrade, L.E.C. Interkit Reproducibility of the Indirect Immunofluorescence Assay on HEp-2 Cells Depends on the Immunofluorescence Reactivity Intensity and Pattern. Front. Immunol. 2021, 12, 798322. [Google Scholar] [CrossRef]
- Thivolet, C.H.; Demidem, A.; Haftek, M.; Durand, A.; Bertrand, J. Structure, function, and immunogenicity of human insulinoma cells. Diabetes 1988, 37, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Beghin, A.; Grenci, G.; Sahni, G.; Guo, S.; Rajendiran, H.; Delaire, T.; Mohamad Raffi, S.B.; Blanc, D.; de Mets, R.; Ong, H.T.; et al. Automated high-speed 3D imaging of organoid cultures with multi-scale phenotypic quantification. Nat. Methods 2022, 19, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Dong, Y.; Chen, T.; Xu, G. Generation of functional fat organoid from rat superficial fascia. Adipocyte 2022, 11, 287–300. [Google Scholar] [CrossRef]
- Hendriks, D.; Artegiani, B.; Hu, H.; Chuva de Sousa Lopes, S.; Clevers, H. Establishment of human fetal hepatocyte organoids and CRISPR-Cas9-based gene knockin and knockout in organoid cultures from human liver. Nat. Protoc. 2021, 16, 182–217. [Google Scholar] [CrossRef]
- Qu, M.; Xiong, L.; Lyu, Y.; Zhang, X.; Shen, J.; Guan, J.; Chai, P.; Lin, Z.; Nie, B.; Li, C.; et al. Establishment of intestinal organoid cultures modeling injury-associated epithelial regeneration. Cell Res. 2021, 31, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Schuth, S.; Le Blanc, S.; Krieger, T.G.; Jabs, J.; Schenk, M.; Giese, N.A.; Buchler, M.W.; Eils, R.; Conrad, C.; Strobel, O. Patient-specific modeling of stroma-mediated chemoresistance of pancreatic cancer using a three-dimensional organoid-fibroblast co-culture system. J. Exp. Clin. Cancer Res. 2022, 41, 312. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Kim, H.J. 3D in vitro morphogenesis of human intestinal epithelium in a gut-on-a-chip or a hybrid chip with a cell culture insert. Nat. Protoc. 2022, 17, 910–939. [Google Scholar] [CrossRef]
- Sasaki, R.; Aoki, S.; Yamato, M.; Uchiyama, H.; Wada, K.; Ogiuchi, H.; Okano, T.; Ando, T. A protocol for immunofluorescence staining of floating neurospheres. Neurosci. Lett. 2010, 479, 126–127. [Google Scholar] [CrossRef]
- Dhar, P.; Das, S.C.; Manu, M.; Mahapatra, C.S.; Latheef, S.K. Development and validation of an in vitro titrimetric method for determination of classical swine fever viruses in PK-15 cells. J. Immunol. Methods 2022, 508, 113321. [Google Scholar] [CrossRef]
- Varani, J.; McClintock, S.D.; Aslam, M.N. Organoid culture to study epithelial cell differentiation and barrier formation in the colon: Bridging the gap between monolayer cell culture and human subject research. Vitr. Cell Dev. Biol. Anim. 2021, 57, 174–190. [Google Scholar] [CrossRef]
- Xia, X.; Li, F.; He, J.; Aji, R.; Gao, D. Organoid technology in cancer precision medicine. Cancer Lett. 2019, 457, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Monleón-Guinot, I.; Milian, L.; Martínez-Vallejo, P.; Sancho-Tello, M.; Llop-Miguel, M.; Galbis, J.M.; Cremades, A.; Carda, C.; Mata, M. Morphological Characterization of Human Lung Cancer Organoids Cultured in Type I Collagen Hydrogels: A Histological Approach. Int. J. Mol. Sci. 2023, 24, 10131. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, J.; Liu, Y.; Li, W.; Chen, G.; Fang, Y.; He, X.; Fu, B.; Jing, Z. A Strainer-Based Platform for the Collection and Immunolabeling of Mouse Intestinal Organoids. Int. J. Mol. Sci. 2023, 24, 13568. https://doi.org/10.3390/ijms241713568
Tan J, Liu Y, Li W, Chen G, Fang Y, He X, Fu B, Jing Z. A Strainer-Based Platform for the Collection and Immunolabeling of Mouse Intestinal Organoids. International Journal of Molecular Sciences. 2023; 24(17):13568. https://doi.org/10.3390/ijms241713568
Chicago/Turabian StyleTan, Jinlong, Yinju Liu, Weike Li, Guohua Chen, Yongxiang Fang, Xiaobing He, Baoquan Fu, and Zhizhong Jing. 2023. "A Strainer-Based Platform for the Collection and Immunolabeling of Mouse Intestinal Organoids" International Journal of Molecular Sciences 24, no. 17: 13568. https://doi.org/10.3390/ijms241713568