
New Insights on the Uptake and Trafficking of Coenzyme Q


Department of Chemistry & Biochemistry and the Molecular Biology Institute, University of California, Los Angeles, CA 90059, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Antioxidants 2023, 12(7), 1391; https://doi.org/10.3390/antiox12071391
Received: 1 June 2023
/
Revised: 30 June 2023
/
Accepted: 30 June 2023
/
Published: 6 July 2023
(This article belongs to the Section Antioxidant Enzyme Systems)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Coenzyme Q (CoQ) is an essential lipid with many cellular functions, such as electron transport for cellular respiration, antioxidant protection, redox homeostasis, and ferroptosis suppression. Deficiencies in CoQ due to aging, genetic disease, or medication can be ameliorated by high-dose supplementation. As such, an understanding of the uptake and transport of CoQ may inform methods of clinical use and identify how to better treat deficiency. Here, we review what is known about the cellular uptake and intracellular distribution of CoQ from yeast, mammalian cell culture, and rodent models, as well as its absorption at the organism level. We discuss the use of these model organisms to probe the mechanisms of uptake and distribution. The literature indicates that CoQ uptake and distribution are multifaceted processes likely to have redundancies in its transport, utilizing the endomembrane system and newly identified proteins that function as lipid transporters. Impairment of the trafficking of either endogenous or exogenous CoQ exerts profound effects on metabolism and stress response. This review also highlights significant gaps in our knowledge of how CoQ is distributed within the cell and suggests future directions of research to better understand this process.
1. Introduction
Coenzyme Q (CoQ), or ubiquinone, is a vital lipid needed for generation of energy and a crucial antioxidant that preserves membrane structure and function. Although it is synthesized within the mitochondria of eukaryotes, it is present in all cellular membranes and is also a component of lipoproteins. CoQ10 (the isoform synthesized by humans and most mammals) is an enormously popular supplement and a medically important therapeutic. Yet, its insolubility in water means it must be trafficked between membranes via lipid vesicles, micelles, chaperones, or transport proteins. The pathways and mechanisms that move this lipid from one membrane to another are still largely undefined. Elucidating the pathways of this trafficking will aid our understanding of the functional roles of CoQ and may lead to improved therapeutics.
In this introductory section, we review the structure and function of CoQ, discuss the genes necessary for its biosynthesis, and provide a short overview of CoQ deficiencies and therapeutic strategies. In the sections that follow, we aim to summarize the work in several model organisms used to investigate how CoQ is trafficked. We will discuss how the yeast Saccharomyces cerevisiae has been used to identify proteins required for the uptake and trafficking of CoQ6 (the isoform of CoQ synthesized by S. cerevisiae), both to and from mitochondria. Mammalian cell culture models have also been used to identify pathways involved in the assimilation of exogenous CoQ10. We will then address how studies in mice, rats, and guinea pigs have been used to identify the uptake and assimilation of exogenous dietary CoQ isoforms.
Human studies and clinical trials addressing the bioavailability and pharmacokinetics of CoQ10 supplements and formulations are the topics of many extensive reviews, and are not discussed here. For readers interested in these topics we recommend the following reviews: [1,2,3,4,5,6,7,8,9]. Additionally, we will not cover the biosynthesis of CoQ in eukaryotic organisms such as Schizosaccharomyces pombe, Arabidopsis thaliana, Caenorhabditis elegans, and in microbes such as Escherichia coli as there are excellent reviews that cover these topics [10,11,12].
1.1. Structure and Function of CoQn
CoQ is a redox-active lipid composed of a fully substituted quinone ring and polyisoprenyl tail of variable length that is species dependent (CoQnwhere n is the number of isoprene units). Humans and most mammals synthesize CoQ10, mice and rats produce CoQ9 with minor amounts of CoQ10, C. elegans CoQ9, E. coli CoQ8, and S. cerevisiae CoQ6 [12,13,14,15]. CoQ can be found in the oxidized state, the semi-reduced state (ubisemiquinone, CoQH·), or the fully reduced state (ubiquinol, CoQH2). CoQ isoforms with polyisoprene tails six units or longer are generally considered to reside at the midplane of the bilayer [16]; however, there are conflicting interpretations regarding the orientation of CoQ isoforms in lipid bilayers [17]. Less hydrophobic isoforms such as CoQ2 and CoQ4 readily diffuse across non-contiguous membranes. Meanwhile longer isoforms, including CoQ6, CoQ9, and CoQ10, are unable to move between lipid vesicles in the absence of a transporter [18,19].
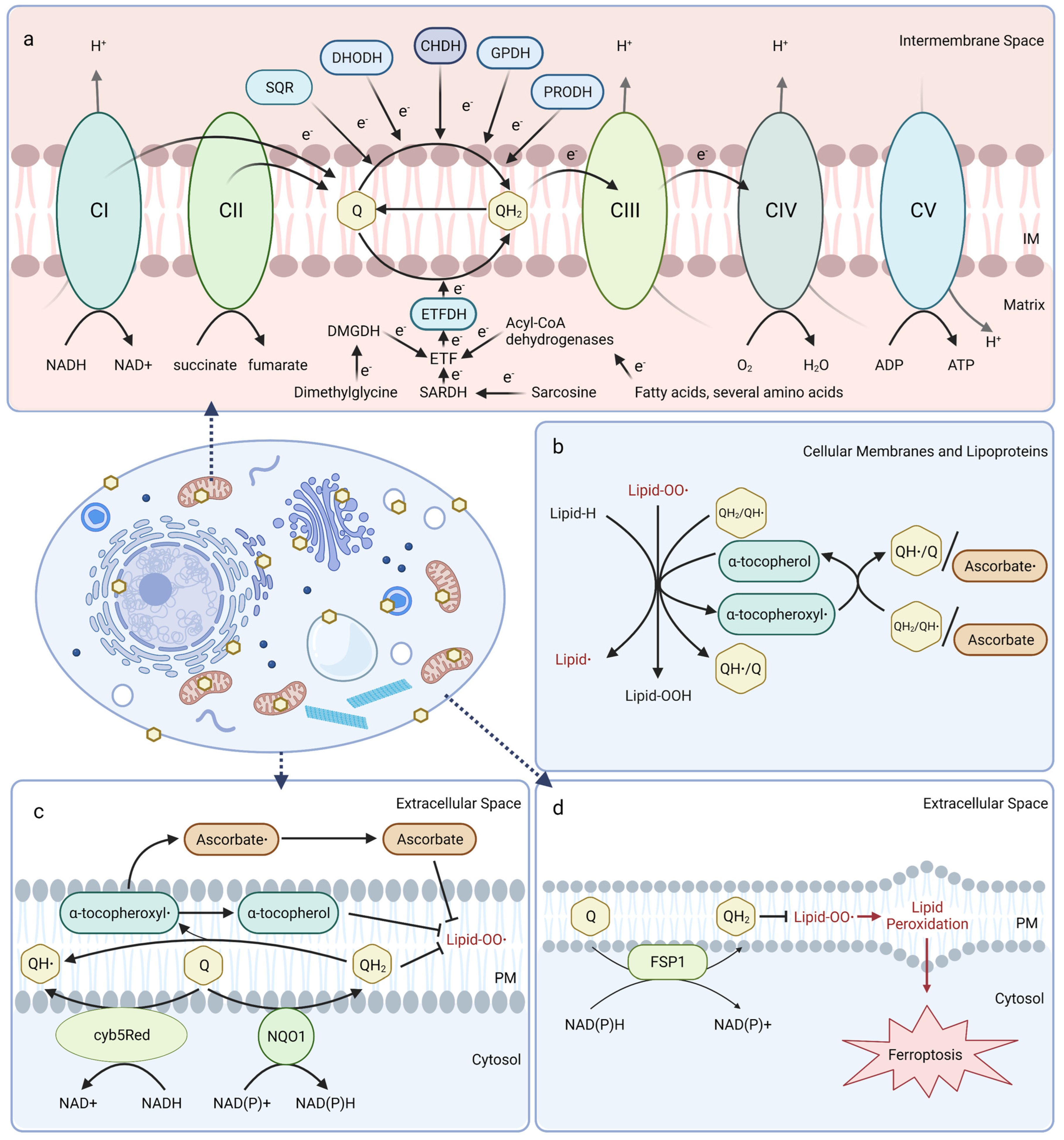
Mitochondrial CoQ plays an essential role in aerobic respiration and can be reduced by oxidoreductases involved in several metabolic pathways (Figure 1a). At the inner mitochondrial membrane, CoQ shuttles electrons along the electron transport chain by accepting electrons from NADH:ubiquinone oxidoreductase (Complex I) and succinate dehydrogenase (Complex II), and donating them to the cytochrome bc1 complex (Complex III). Cytochrome c then transports electrons from Complex III to cytochrome oxidase (Complex IV), which directly reduces O2 into water. The free energy from this electron transfer is used to transport protons from the matrix side to the intermembrane space, ultimately creating a proton-motive force used by ATP synthase to generate ATP [20,21,22]. Mitochondrial CoQ is also reduced by several other dehydrogenases and thus participates in diverse metabolic pathways, including sulfide:quinone oxidoreductase (SQR) for catabolism of sulfide, dihydroorotate dehydrogenase (DHODH) for pyrimidine biosynthesis, choline dehydrogenase (CHDH) for choline oxidation, glycerol-3-phosphate dehydrogenase (GPDH) for the glycerol-3-phosphate shuttle, proline dehydrogenase (PRODH) for catabolism of proline, and electron-transferring-flavoprotein dehydrogenase (ETFDH) for oxidation of fatty acids, branched-chain amino acids, sarcosine, and dimethylglycine (Figure 1a) [23].
In mitochondrial and other biological membranes, CoQ functions as a powerful antioxidant and plays a role in cell maintenance. CoQ is the only endogenously synthesized lipid-soluble antioxidant and acts to inhibit lipid peroxidation from damaging cellular membranes, proteins, and DNA. CoQH2 acts as a chain terminator to inhibit both the initiation and propagation steps of autoxidation of polyunsaturated fatty acids (PUFAs) [24,25]. Moreover, CoQH2 is also capable of regenerating α-tocopherol (vitamin E) from the tocopheroxyl radical (Figure 1b) [26]. Recent studies indicate that the oxidized form of CoQ can also slow peroxyl radical addition reactions responsible for cellular damage incurred by electrophilic stress resulting from conjugated PUFAs, such as conjugated linoleic and conjugated linolenic acids [27].
CoQ at the plasma membrane is part of the plasma membrane redox system (PMRS), which combats oxidative stress, maintains cell bioenergetics when mitochondrial activity decreases, and is involved in cell growth and resistance to apoptosis (Figure 1c) [28,29]. The PMRS comprises NAD(P)H:quinone oxidoreductase 1 (NQO1), NADH-cytochrome b5 reductase (cyb5Red), CoQ, and other antioxidants, such as vitamin E and ascorbate. The PMRS functions to regulate cellular redox homeostasis through the maintenance of cytosolic NAD(P)+/NAD(P)H and CoQ/CoQH2 pools within the plasma membrane. CoQH2 generated by the PMRS can function as a non-competitive inhibitor of neutral sphingomyelinase, a membrane protein that releases ceramide from membrane sphingomyelin to activate ceramide-dependent caspases and elicit apoptosis in serum-deprived cells [29,30,31]. Additionally, ferroptosis suppressor protein 1 (FSP1) was recently identified as a CoQ reductase that regenerates CoQH2 at the plasma membrane to inhibit ferroptosis in a pathway that is distinct from the anti-ferroptotic glutathione peroxidase 4 (GPX4) system (Figure 1d) [32,33]. Intriguingly, vitamin K is also reduced by FSP1 and inhibits ferroptosis [34], while tetrahydrobiopterin may function as a co-antioxidant with CoQ to inhibit ferroptosis [35]
1.2. CoQ Biosynthesis, Mutations in COQ Genes, and Therapeutic Strategies
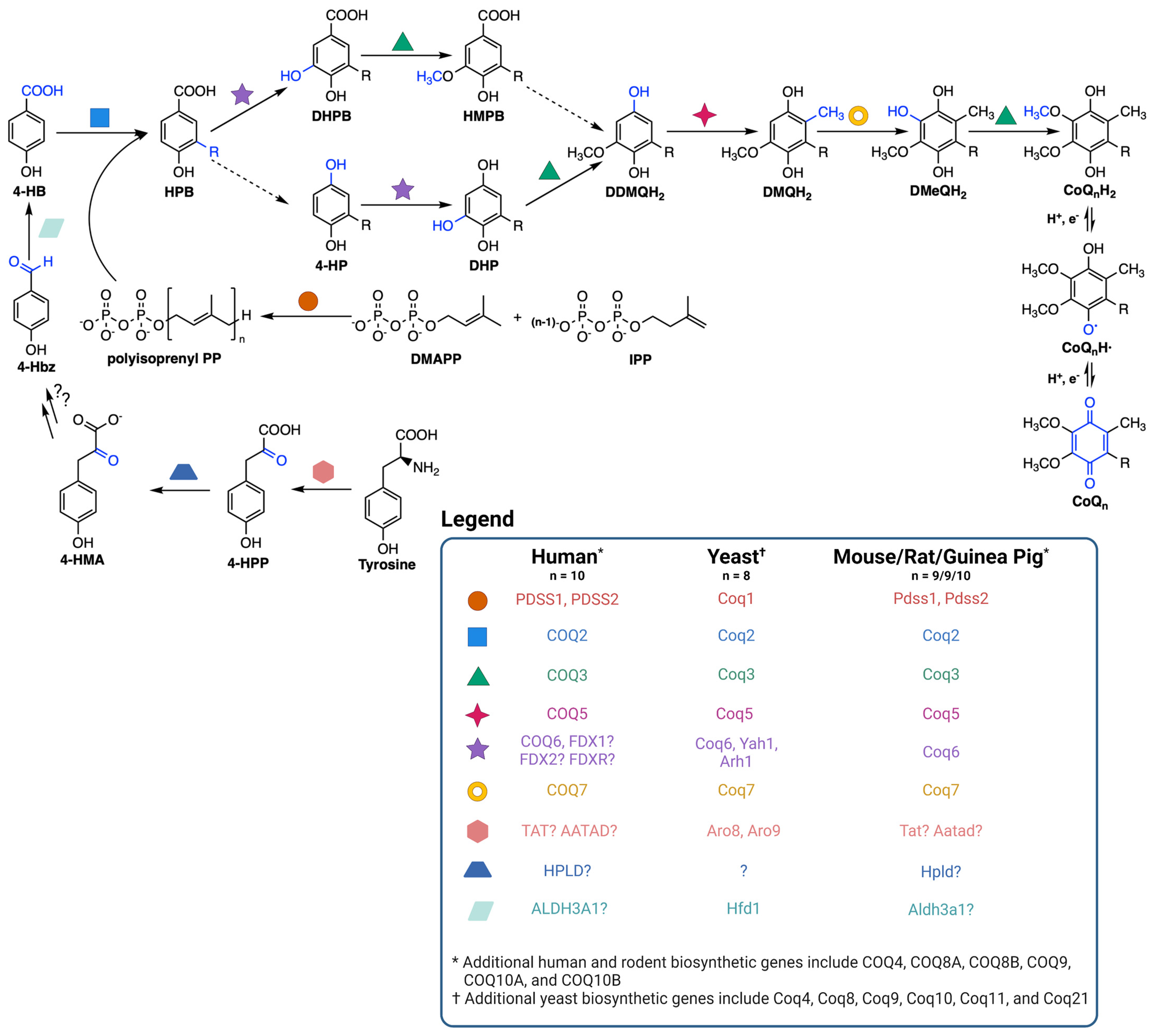
CoQ is synthesized at the inner mitochondrial membrane on the matrix side, but is found throughout all cellular membranes [36]. CoQ biosynthesis requires a high molecular mass complex, termed the CoQ synthome in yeast and complex Q in mammals, situated on the matrix side of the inner mitochondrial membrane (Figure 2) [37,38,39]. CoQ biosynthesis begins with formation of the aromatic ring precursor 4-hydroxybenzoic acid (4-HB). In yeast, 4-HB is derived from the shikimate pathway or from tyrosine [38]. Human cells, however, may derive 4-HB from phenylalanine or tyrosine due to the presence of phenylalanine hydroxylase [37]. Deamination of tyrosine produces 4-hydroxyphenylpyruvate (4-HPP). In humans, mouse, rat, and guinea pig, this step is thought to be catalyzed by tyrosine aminotransferase (TAT) or by mitochondrial alpha-aminoadipate aminotransferase (AATAD) [37]. In yeast there is considerable redundancy in the pathway of 4-HB synthesis; five aminotransferase enzymes can perform this deamination step [40]. In humans, hydroxyphenylpyruvate dioxygenase-like protein (HPLD) converts 4-HPP to 4-hydroxymandelate (4-HMA) [41]. 4-HMA is also a likely intermediate in the yeast pathway to 4-HB [40,42]. The additional steps producing 4-hydroxybenzaldehyde (4-Hbz) have yet to be determined in either yeast or human cells. In yeast, Hfd1 is required to oxidize 4-Hbz to 4-HB, and the human aldehyde dehydrogenase 3A1 (ALDH3A1) has also been shown to perform this step [37]. COQ2, or Coq2 in yeast, first attaches the variable length polyisoprenyl tail to 4-HB. The polyprenylated intermediate is then introduced to the CoQ synthome (or complex Q), where the COQ3-COQ9 polypeptides act in concert to synthesize CoQ (Figure 2) [10,38]. The final CoQ product is then transported to its intracellular destination in the mitochondria, plasma membrane, or other organellar membranes via poorly characterized pathways [37].
Mutations in the CoQ biosynthetic genes may result in a condition termed primary CoQ10 deficiency. Primary CoQ10 deficiency is a rare disease often associated with multisystem disorders [43]. However, deleterious mutations that produce a poorly functioning or non-functional polypeptide, can also result in isolated phenotypic consequences, affecting the kidney or central nervous system. Genetic or biochemical testing is required for comprehensive diagnosis [43,44]. Secondary CoQ10 deficiency may arise when mutations in genes not directly associated with CoQ10 biosynthesis affect cellular CoQ10 content [45], in response to drugs (such as statins), or as a result of aging [39]. Currently, the only treatment for CoQ10 deficiency is high-dose exogenous supplementation of CoQ10. While CoQ10 displays poor bioavailability, in some patients presenting with primary CoQ10 deficiency supplementation produced dramatic improvements in their long-term clinical phenotypes [6,46]. To improve such clinical outcomes, it is important to understand both the gastrointestinal (GI) uptake pathway of orally supplemented CoQ10, as well as the cellular uptake and intracellular distribution.
Figure 2.
Coenzyme Q biosynthetic pathway. The dashed arrows indicate decarboxylation and hydroxylation steps that are catalyzed by unknown enzyme(s), thus resulting in an uncertainty in the order of reactions. Intermediates in the pathway include 4-HPP, 4-hydroxyphenylpyruvate; 4-HMA, 4-hydroxymandelate; 4-Hbz, 4-hydroxybenzaldehyde; 4-HB, 4-hydroxybenzoic acid; DMAPP, dimethylallyl pyrophosphate; IPP, isopentenyl pyrophosphate; HPB, 4-hydroxy-3-polyprenyl-benzoic acid; DHPB, 4,5-dihydroxy-3-polyprenylbenzoic acid; HMPB, 4-hydroxy-5-methoxy-3-polyprenylbenzoic acid; DHP, 4,5-dihydroxy-3-polyprenylphenol; DDMQH2, 2-methoxy-6-polyprenyl-1,4-benzohydroquinone; DMQH2, 2-methoxy-5-methyl-6-polyprenyl-1,4-benzohydroquinone; DMeQH2, 3-methyl-6-methoxy-2-polyprenyl-1,4,5-benzenetriol; n is the number of isoprene units in the polyisoprenyl tail. Yeast can also utilize p-aminobenzoic acid as a ring precursor, and its prenylation by Coq2 produces the early intermediate 4-amino-3-polyprenyl-benzoic acid (HAB, [38]). Adapted with permission from Wang et al. [44]. Created with Biorender.com (accessed on 28 June 2023).
Figure 2.
Coenzyme Q biosynthetic pathway. The dashed arrows indicate decarboxylation and hydroxylation steps that are catalyzed by unknown enzyme(s), thus resulting in an uncertainty in the order of reactions. Intermediates in the pathway include 4-HPP, 4-hydroxyphenylpyruvate; 4-HMA, 4-hydroxymandelate; 4-Hbz, 4-hydroxybenzaldehyde; 4-HB, 4-hydroxybenzoic acid; DMAPP, dimethylallyl pyrophosphate; IPP, isopentenyl pyrophosphate; HPB, 4-hydroxy-3-polyprenyl-benzoic acid; DHPB, 4,5-dihydroxy-3-polyprenylbenzoic acid; HMPB, 4-hydroxy-5-methoxy-3-polyprenylbenzoic acid; DHP, 4,5-dihydroxy-3-polyprenylphenol; DDMQH2, 2-methoxy-6-polyprenyl-1,4-benzohydroquinone; DMQH2, 2-methoxy-5-methyl-6-polyprenyl-1,4-benzohydroquinone; DMeQH2, 3-methyl-6-methoxy-2-polyprenyl-1,4,5-benzenetriol; n is the number of isoprene units in the polyisoprenyl tail. Yeast can also utilize p-aminobenzoic acid as a ring precursor, and its prenylation by Coq2 produces the early intermediate 4-amino-3-polyprenyl-benzoic acid (HAB, [38]). Adapted with permission from Wang et al. [44]. Created with Biorender.com (accessed on 28 June 2023).

The rescue of CoQ10-deficient animals supplemented with synthetic hydrophilic CoQ precursors or “bypass therapies” indicates that bioavailability is a key limitation of CoQ10 supplementation [6]. Bypass therapy for patients with primary CoQ deficiency is currently being studied as a potential alternative to supplementation with CoQ10 [47,48]. In bypass therapies, analogs of 4-HB are used as alternate ring precursors that function to “bypass” a blocked step in the CoQ biosynthetic pathway. For example, 2,4-dihydroxy-benzoic acid (2,4-diHB) partially restores CoQ biosynthesis in mice harboring mutations in Coq7 (also termed Mclk1) [49]. Mice with certain mutations of Coq9 also respond to 2,4-diHB treatment [50]. However, 2,4-diHB also inhibits the endogenous CoQ biosynthetic pathway, possibly due to competition with natural substrates of the COQ enzymes upstream of the Coq7 hydroxylation step [49]. Bypass of human COQ6 mutations using vanillic acid, a methoxylated analog of 4-HB, also has efficacy in a cultured cell model [51]. Due to the lack of a hydrophobic polyisoprenyl tail, the analogs of 4-HB are significantly more bioavailable than CoQ10, and the restoration of endogenous biosynthesis may achieve a more effective rescue [52].
2. The S. cerevisiae Model
S. cerevisiae (hereafter referred to as yeast) is a single-celled fungus that has been widely studied as a simple, eukaryotic model organism. Yeast cells are capable of both fermentation and aerobic respiration, and they divide rapidly through a budding process in which smaller daughter cells bud off from a larger mother cell. These cells are approximately five microns in diameter, between bacterial and human cells in size. Yeast cells can readily switch between haploid a and haploid α cells and mate to form diploid cells, which can further sporulate to produce four haploids [53]. Many eukaryotic cellular processes have been first identified and extensively characterized in yeast, including cell cycle regulation, vesicle trafficking, protein secretion, and autophagy. Many of these processes are highly conserved between yeast and humans. In fact, 47% of the 414 essential genes in S. cerevisiae with a 1:1 human ortholog can be functionally replaced by the corresponding human ortholog [54]. These well-characterized pathways, ease of genetic manipulation, and strong conservation between yeast and humans make yeast a powerful model organism.
Much work has been performed to elucidate the biosynthetic pathway and understand the cellular function of CoQ6 in yeast. The COQ1–COQ11 genes encode mitochondrially targeted polypeptides that are required for efficient CoQ6 biosynthesis [37,38,39]. Strains with a deletion or deleterious mutation in one of the COQ1–COQ9 genes, termed coq mutants, are unable to synthesize CoQ6 (Figure 3a). These coq mutants are thus incapable of cellular respiration and fail to grow in a non-fermentable medium. In most cases, expression of the human COQ ortholog in the respective yeast coq mutant rescues efficient CoQ6 biosynthesis and restores growth in a non-fermentable medium, highlighting the strong conservation of CoQ biosynthesis from yeast to humans. In addition to the presence of all the Coq polypeptides, efficient CoQ6 synthesis requires Coq3–Coq9 and Coq11 to form a mega-complex on the matrix side of the inner mitochondrial membrane, termed the CoQ synthome [38,39]. The formation of this complex is analogous to the Complex Q assembly that occurs in human cells [39]. The strong conservation of CoQ biosynthesis and many functions from yeast to humans suggest a conserved pathway of CoQ transport.
2.1. Growth Rescue of coq Mutants in a Non-Fermentable Medium Supplemented with Exogenous CoQ6
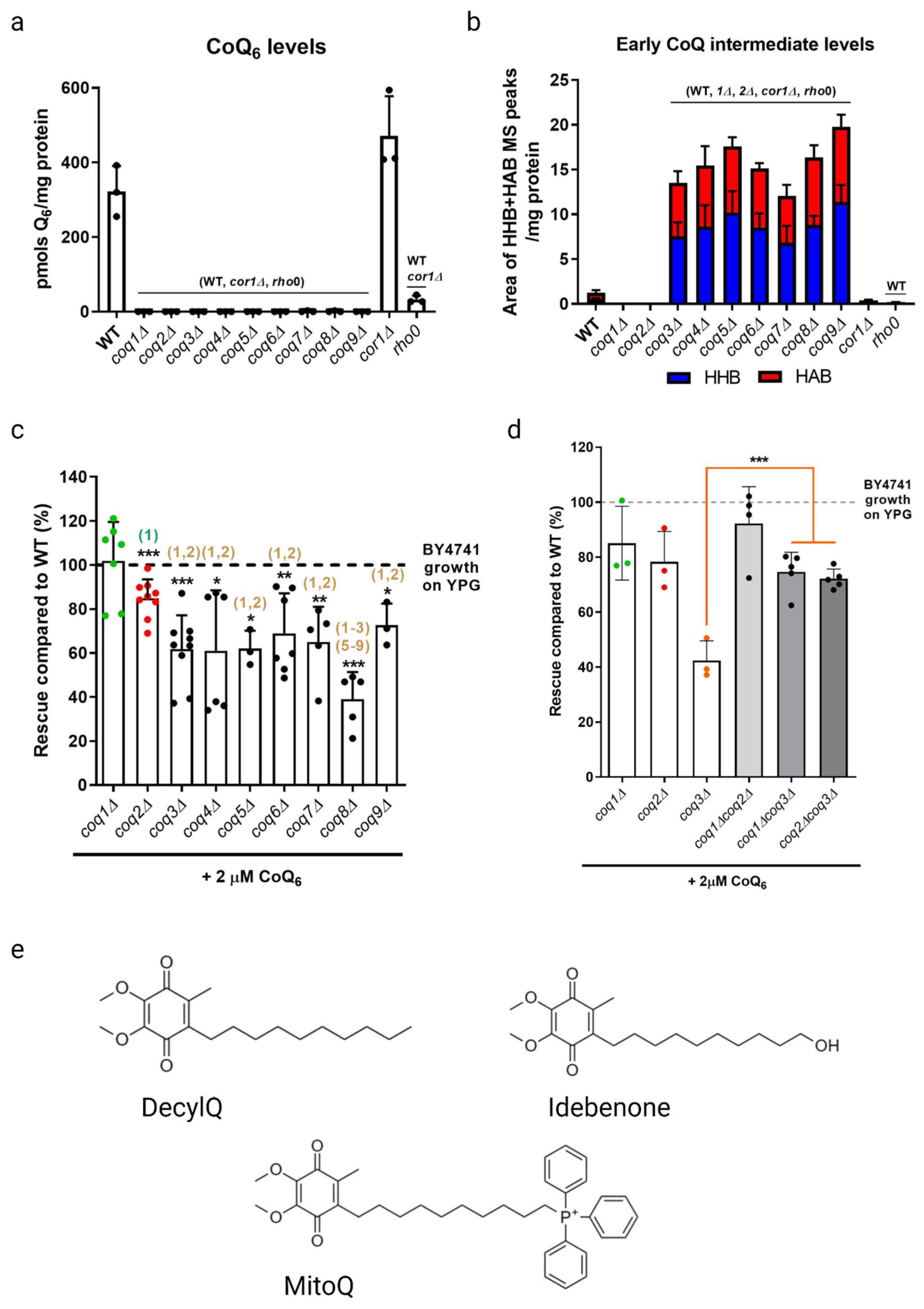
As previously stated, coq mutants are unable to synthesize CoQ6 and are thus respiratory deficient and fail to grow in a medium containing a non-fermentable carbon source (Figure 3a). Deletion of COQ1 or COQ2 that are involved in early steps of CoQ6 biosynthesis prevents the accumulation of early CoQ6 intermediates, 3-hexaprenyl-4-hydroxybenzoic acid (HHB) and 3-hexaprenyl-4-aminobenzoic acid (HAB), while deletion of any one of the COQ3-COQ9 genes that are required for later biosynthetic steps accumulate these early CoQ intermediates (Figure 3b). Significantly, direct supplementation of exogenous CoQ6 restores respiration and growth of coq mutants [55,56], though the degree of rescue depends on the nature of the coq mutant. When supplemented with exogenous CoQ6 in non-fermentable media, coq1Δ and coq2Δ single mutants exhibit wild-type or near wild-type levels of growth, while each of the coq3Δ–coq9Δ single mutants exhibit growth that is significantly lower than wild-type cells (Figure 3c). Deletion of either COQ1 or COQ2 in the coq3Δ single mutant restores rescue to that of wild-type cells or to the coq1Δ and coq2Δ single mutants (Figure 3d), indicating that the accumulation of early CoQ6 intermediates, HHB and HAB, impairs growth rescue of coq3Δ–coq9Δ mutants [18]. It is possible that HHB/HAB act as detergents or may interfere with membrane trafficking steps needed for transit of exogenously added CoQ6. CoQ6 uptake is favored by the presence of peptone in the culture media [57]. The digested proteins in peptone likely bind CoQ6 and enable its entry into the aqueous compartment of endocytic vesicles. Following supplementation with exogenous CoQ6, the coq mutants also show a significant delay in growth compared to wild-type cells as they make the diauxic shift from fermentation to respiration [57]. Supplementation with either hydrophilic CoQ isoforms that can spontaneously diffuse across non-continuous membranes (CoQ2, CoQ4), or hydrophobic CoQ isoforms that cannot diffuse (CoQ6) are both capable of rescuing growth. Moreover, supplementation with CoQ2 can bypass defects in endocytosis and membrane trafficking [19,58]. The ability of yeast to uptake exogenous CoQ, transport it to the mitochondria, and in turn restore respiration in coq mutants makes yeast a powerful model organism to investigate CoQ uptake and transport.
Studies with supplementation of non-isoprenoid CoQ analogs indicate that the isoprenoid tail is essential for efficient uptake and growth rescue of coq mutants in a non-fermentable medium. Idebenone, decylQ, and MitoQ are three such analogs containing a ten-carbon alkyl tail (Figure 3e). The tail end of idebenone and MitoQ are further modified with a hydroxyl group or triphenylphosphonium moiety, respectively, with the latter exhibiting mitochondrial targeting. None of these three analogs were able to restore growth of coq mutants when supplemented in a non-fermentable medium [58]. Although decylQ and idebenone restore respiration in isolated mitochondria, it is speculated that mitochondrial accumulation of these analogs in whole cells is insufficient to support growth. Conversely, MitoQ accumulates in mitochondria of whole cells, but is poorly oxidized by Complex III and thus unable to restore respiration [58]. In fact, supplementation with high concentrations of decylQ inhibits growth rescue of coq mutants with CoQ2, perhaps through competition with an unidentified CoQ-binding protein [19]. Supplementation assays with analogs of CoQ2 where the prenyl tail has been modified show that each of the analogs tested are capable of restoring respiration in isolated mitochondria, but the presence of the double bond between C2 and C3 in the first isoprenoid unit of exogenously supplied CoQ is essential for efficient uptake in whole cells and growth in a non-fermentable medium [19]. Taken together, the decylQ impairment of growth rescue by CoQ2 and the structural requirement in the isoprenoid tail of exogenous CoQ suggest the presence of an essential protein that can distinguish subtle differences in CoQ structure for efficient delivery or retention of exogenous CoQ to mitochondrial respiratory complexes.
It is possible that the yeast Coq10 polypeptide may fulfill this role of a CoQ chaperone. Yeast Coq10 is peripherally associated with the matrix side of the inner mitochondrial membrane, but is not part of the endocytic pathway, the respiratory complexes, or the CoQ synthome [59]. The yeast Coq10 polypeptide contains a steroidogenic acute regulatory-related lipid transfer (START) domain, which binds CoQn isoforms of variable tail lengths and analogs of late-stage CoQ-intermediates such as DMQ3 [60]. Yeast Coq10 is needed for respiratory function, and may be involved in transport of newly synthesized CoQ6 to Complex III [61]. COQ10A and COQ10B are human orthologs of yeast Coq10, and partially rescue both the respiratory defect and sensitivity to lipid peroxidation of the coq10Δ yeast mutant [62]. Human STARD7 is structurally related to the yeast Coq10 and human COQ10 polypeptides, binds CoQ4 in vitro, and plays a role in vivo in transporting mitochondrial CoQ10 to the plasma membrane to suppress ferroptosis [63]. Human saposin B binds CoQ10, and is postulated to function as a transporter [64,65]. Mammalian CoQ transport proteins are discussed in Section 3.1.5. It is likely that many other CoQ transporter or chaperone proteins remain to be discovered.
2.2. Identification of Essential Genes in CoQ6 Uptake and Trafficking to Mitochondria Respiratory Complexes with the Use of Yeast ORFΔcoqΔ Double Mutants
2.2.1. A Screen for Genes That Impact Growth Rescue by Exogenous CoQ
Previous studies performed by James et al. have sought to identify genes that influence the ability of exogenously supplemented CoQ2 and CoQ4 to support the respiratory growth of CoQ-less yeast [19]. Sixteen such genes were found by screening a library of ~4800 ORFΔcoqΔ double mutants. The deletion of these ORFs resulted in impaired growth rescue on a medium containing a non-fermentable carbon source when supplemented with exogenous CoQ2 or CoQ4. However, it is important to note that CoQ2 and CoQ4 have shorter isoprenoid tails than CoQ6, passively diffuse across noncontinuous membranes, and move independently of lipid trafficking mechanisms [18,19].
To further examine the role of these ORFs as well as other candidates in the uptake and trafficking of CoQ6, Fernández-Del-Río et al. tested whether exogenous CoQ6 restored growth in a screen of 40 ORFΔcoq2Δ double mutants in a non-fermentable medium [18]. This group of 40 genes included the 16 ORFΔcoq2Δ double mutants identified by James et al. [19]. The rescue assay compared the ORFΔcoq2Δ double mutant to the coq2Δ single mutant, and to the ORFΔ single mutant in a non-fermentable medium when supplemented with exogenous CoQ6 [18]. The degree of growth rescue in a medium supplemented with exogenous CoQ6 was shown to be dependent on the distinct COQ gene that was disrupted (see Section 2.1). Six ORFΔcoq2Δ double mutants demonstrated reduced growth in a non-fermentable medium with exogenous CoQ6 as compared to either the coq2Δ or the ORFΔ single mutants, suggesting a role for these ORFs in CoQ6 uptake and transport to the mitochondria. Fernández-Del-Río et al. reasoned that supplementation with the hydrophilic isoform CoQ2 should be able to circumvent the trafficking defect observed with CoQ6 in these mutants [18]. Indeed, the growth rescue afforded by CoQ2 supplementation in these ORFΔcoq2Δ double mutants far exceeded that of CoQ6. Such growth rescue assays of ORFΔcoq2Δ double mutants provide a powerful tool to identify genes that are required for the uptake of exogenous CoQ6 and its trafficking to mitochondria. However, more work is needed to unravel the role these gene products play in uptake and transport of exogenous CoQ6.
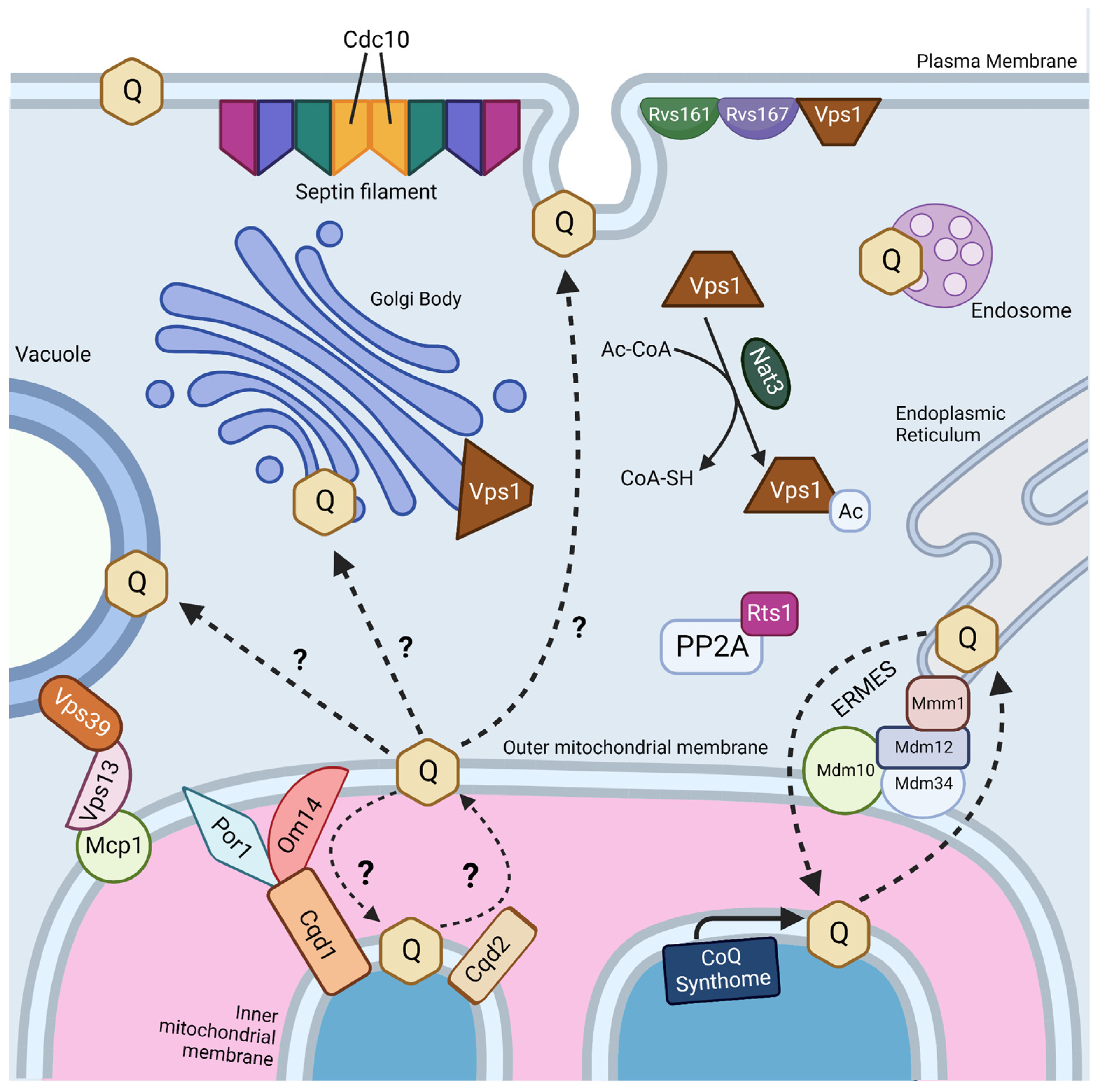
Interestingly, two genes, RTS1 and CDC10, were identified as essential in both screens (Figure 4) [18,19]. Rts1 is a regulatory subunit of protein phosphatase 2A (PP2A), which plays a role in regulation of cell size control, the mitotic spindle orientation checkpoint, and septin ring organization and disassembly during cytokinesis [66,67]. It is possible that Rts1 in complex with PP2A plays a regulatory role in CoQ6 uptake and transport. CDC10 is one of four core septins in yeast, a group of GTPases that assembles into filaments and higher order structures [68]. Septin filaments assemble into a septin ring at the bud neck in yeast and act as a scaffold to recruit other proteins to drive cytokinesis [69]. Septins have been shown to interact with a subset of endocytic proteins [70], and SEPT9, the human homolog of Cdc10, has been implicated in endosomal trafficking [71]. More work is needed to determine if the role of Cdc10 in CoQ6 uptake and transport is involved in endocytosis or another septin function.
The four other essential genes for uptake and trafficking of exogenous CoQ6 to respiratory complexes identified by Fernández-Del-Río et al. are RVS161, RVS167, NAT3, and VPS1 (Figure 4) [18]. Rvs161 and Rvs167 are amphiphysin-like membrane proteins that form a heterologous dimer and promote membrane curvature [72]. Rvs161 and Rvs167 interact with several other proteins, including Vps1, at the plasma membrane for the vesicle scission step of endocytosis [73]. Vps1 is a GTPase involved in membrane fusion and fission. Vps1 also operates in other cellular processes, including vacuolar protein sorting, actin cytoskeleton organization, late Golgi retention of proteins, and peroxisome biogenesis [74]. Nat3 is a catalytic subunit of the NatB complex that mediates acetylation of the N-terminus of approximately 20% of proteins in yeast and humans [75,76]. The role that each of these genes plays in CoQ6 uptake and trafficking remains unclear.
Typically, mitochondria are excluded from schemes depicting endosomal trafficking [77]. This is curious because trafficking between the plasma membrane and mitochondria is reported to occur via clathrin-mediated endocytosis [78]. However, trafficking of proteins and lipids to mitochondria is likely mediated by extensive contact sites between mitochondria and other intracellular membranes (Figure 4) [79,80]. Fernández-Del-Rio et al. screened ORFΔcoq2Δ double mutants predicted to impair selected membrane contact sites such as the endoplasmic reticulum-mitochondria encounter structure (ERMES), the nucleus-vacuole junction (NVJ), and the vacuole–mitochondria patch (vCLAMP) [18].
However, none of these ORFΔcoq2Δ double mutants displayed impaired growth rescue when treated with exogenous CoQ6. These results could suggest that CoQ6 transport may occur independently of these membrane contact sites, or that transport through contact sites is redundant. For example, mitochondria dependence on phospholipid exchange has been shown to depend on either ERMES or vCLAMP [81]. Loss of ERMES stimulates vCLAMP and vice versa; however, loss of both is lethal [81]. Yeast genes that are redundant or essential for growth on a non-fermentable carbon source will not be detected by such exogenous CoQ6 growth-rescue screens. It is also possible that other lipid transport mechanisms operate independently of both endocytic and contact site-mediated trafficking.
2.2.2. Rescue of coq Mutants with Exogenous CoQ Is Variable and Depends on the Genetic Background of the Yeast Strain
Growth rescue of coq mutants in a non-fermentable medium with exogenous CoQ6 is dependent on the yeast genetic background. For example, the commonly used BY4741/BY4742 laboratory yeast strain takes up and transports exogenous CoQ6 to the mitochondrial respiratory complexes, and was used in the screen to detect trafficking defects (Section 2.2.1) [18]. This is also the case for the W303-1B and CEN.PK2-1C yeast genetic backgrounds [82]. Yet, certain yeast strains harboring a coq7Δ null allele, such as the EG103coq7Δ or the FY250coq7Δ mutant yeast strains, fail to be rescued when the growth medium is supplemented with exogenous CoQ6. The content of CoQ6 in the gradient-purified mitochondrial fraction of these strains was only 35% and 8% of the content present in the mitochondria isolated from the EG103 and FY250 wild-type parental strains, respectively. However, the CoQ6 content in the plasma membrane fraction isolated from EG103coq7Δ and the FY250coq7Δ strains exceeded or equaled the content of CoQ6 normally present in the plasma membrane fraction of the parental strains [82]. In these mutant strains, the plasma membrane redox activity was augmented, suggesting that the exogenous CoQ6 present in the plasma membrane was functional [82]. Thus, it is likely that such strains unable to be rescued for growth by exogenous CoQ6 likely have a defect in trafficking the CoQ6 from the plasma membrane to the mitochondria [82]. Both the parental EG103 and the EG103coq7Δ mutant strains were shown to secrete carboxy-peptidase Y and accumulate small vacuoles, markers that characterize impaired membrane trafficking [57]. It would be important to test whether these mutants are rescued by supplementation with exogenous CoQ2, since this analog does not depend on lipid trafficking.
To test the idea that defects in endocytosis or endomembrane trafficking alter CoQ6 uptake and transport to the mitochondria, Padilla-López et al. interrogated ORFΔcoq3Δ double mutants whose ORF deletion, such as ERG2, PEP12, TLG2, or VPS45, disrupts several endocytic transport steps [57]. Supplementation with exogenous CoQ6 failed to rescue growth of these ORFΔcoq3Δ double mutants in a non-fermentable medium, suggesting that the endocytic and endomembrane trafficking pathways are involved in CoQ6 uptake and transport. Subcellular fractionation confirmed that each of these ORFΔcoq3Δ double mutants have decreased uptake of exogenous CoQ6 in mitochondria, Golgi, vacuole, and plasma membrane compared to WT and coq3Δ controls, further illustrating the impact of disrupting these trafficking pathways in CoQ6 uptake. Moreover, the low CoQ6 content in the plasma membrane of these mutants suggests that most of the exogenously supplied CoQ6 cannot be directly incorporated into the plasma membrane.
It must be noted that both coq7Δ and coq3Δ mutants accumulate HHB and HAB, early CoQ6 intermediates that act to impair growth rescue in a non-fermentable medium with exogenous CoQ6. Indeed, Fernández-Del-Río et al. showed that these ORFΔcoq3Δ double mutants had significantly less robust growth in a non-fermentable medium when compared to the ORFΔcoq2Δ double mutant counterpart (Figure 3c,d) [18]. More work must be performed in yeast to further determine the role of endocytosis and endomembrane trafficking in CoQ6 uptake and transport in yeast models. Previous work in the human promyelocytic HL-60 cell line has shown that both endogenously synthesized CoQ10 and exogenously supplied CoQ9 are distributed across membranes through the brefeldin A-sensitive endo-exocytic pathway (see Section 3.1.1) [83]; however, such experiments have not been performed in yeast.
2.3. CoQ6 Transport to and from the Mitochondria
It is likely that uptake and trafficking of exogenous and endogenous CoQ6 share common pathways. Recent studies suggest that the ERMES may play a role in both CoQ6 biosynthesis and distribution. ERMES is a tethering complex that forms at the membrane contact site between the ER and the outer mitochondrial membrane and is composed of two mitochondrial subunits embedded in the outer membrane (Mdm10 and Mdm34), an ER-localized subunit (Mmm1), and a cytosolic subunit (Mdm12) (Figure 4) [84]. The four subunits are functionally linked to phospholipid biosynthesis and calcium signaling [84]. Recent structural analyses show that ERMES forms bridge-like structures between the ER and mitochondrial outer membrane, consistent with providing a conduit for phospholipid transport [85]. The mitochondrial contact site and cristae organization system (MICOS), a mega complex that regulates cristae junction organization, has been shown to assemble near ERMES sites, and ERMES function is required for MICOS assembly [86]. The Mmm1-Mdm12 complex has been shown to mediate phospholipid transfer in vitro and mutations in MMM1 and MDM12 lead to impaired lipid transfer through ERMES in vivo [87].
Intriguingly, Coq polypeptides that are components of the CoQ synthome at the inner mitochondrial membrane are visualized as puncta or CoQ domains that lie adjacent to ERMES puncta at the outer mitochondrial membrane [88,89]. Disruption of ERMES decreases the number of CoQ domains [88,89]. Moreover, ERMES mutants show increased whole-cell accumulation of CoQ6 and CoQ6-intermediates but decreased content of CoQ6 in isolated mitochondria, suggesting that CoQ6 and CoQ6-intermediates accumulate in non-mitochondrial membranes [89]. Current observations suggest that destabilization of the CoQ synthome results in reduced sequestration of CoQ6 and CoQ6 intermediates in the mitochondria; alternatively, disruption of ERMES might lead to enhanced stability of non-mitochondrial CoQ6 [89]. Despite this role for ERMES in CoQ domain formation and CoQ6 production and distribution, direct transport of CoQ6 through ERMES has not yet been observed. Moreover, none of the four ERMES genes were identified as essential for CoQ uptake and transport in ORFΔcoq2Δ double mutants [18]. However, disruption of ERMES results in an expansion of vCLAMP contact sites, and deletion of both contacts has been shown to be lethal [81]. Thus, disruption of ERMES might increase compensatory contact sites to counteract potential defects in CoQ6 trafficking.
The idea that other membrane contact sites can compensate for lipid transport defects in ERMES mutants has been reinforced by recent structure/function studies of the VPS13 family of proteins. Yeast Vps13 is a 360 kDa protein composed of an extensive beta-sheet or “taco shell” lined by hydrophobic residues that can form a conduit for phospholipid transfer between organelles [90]. Yeast Vps13, Vps39, and Mcp1 form a mitochondria–vacuole contact site (Figure 4), and are required for the survival of yeast ERMES mutants [91]. Mcp1 is a mitochondrial outer membrane phospholipid scramblase, and partners with the Vps13 lipid bridge protein to re-equilibrate lipids as they are either inserted into or removed from the cytosolic leaflet of acceptor or donor membranes [92]. Vps39 is the vacuole fusion factor responsible for vCLAMP contact sites [81]. It is not yet known whether these bridge-like phospholipid transporters strategically positioned at contact sites also move neutral lipids, such as CoQ.
Recent work has identified yeast mitochondrial Cqd1 and Cqd2 proteins that influence cellular distribution of endogenous CoQ6 [93]. Intriguingly, both are inner membrane proteins that face the intermembrane space, and potentially provide a connection for transfer of CoQ6 from the inner membrane to the outer membrane and the ER (Figure 4). Disruption of CQD1 or CQD2 either diminishes or enhances the mitochondrial CoQ6 content, respectively, without altering overall cellular CoQ6 content [93]. Cqd1 and Cqd2 are members of the UbiB family of atypical kinases/ATPases. Coq8, an essential protein for CoQ6 biosynthesis and part of the CoQ synthome, is also a member of the UbiB family whose ATPase activity may be coupled to the extraction of hydrophobic CoQ6 intermediates from the inner membrane for subsequent processing by membrane-associated Coq polypeptides in the matrix [94]. Point mutations of both Cqd1 and Cqd2 at conserved protein-kinase-like (PKL) or UbiB motif residues disrupted their individual impact on CoQ6 distribution, indicating that Cqd1 and Cqd2 activities rely on atypical kinase/ATPase activity, though further biochemical work is needed to prove enzymatic activity. Furthermore, haploinsufficiency of the CQD1 human ortholog ADCK2 is reported to cause aberrant mitochondrial lipid oxidation and myopathy associated with CoQ10 deficiency [95].
Cqd1 participates in a contact site between outer and inner mitochondrial membranes (Figure 4), and overexpression of Cqd1 and Cqd2 elicits contact sites between the ER and mitochondria [96]. Taken in conjunction with the effect of Cqd1 on CoQ6 retention within mitochondria, and the effect of Cqd2 on promoting CoQ6 exit from mitochondria, these proteins likely play a role in CoQ6 trafficking to and from the mitochondria in yeast. It would be interesting to determine whether the Cqd1 complex is co-localized with the CoQ synthome and ERMES, as it provides the “missing link” between these two. Interestingly, deletion of both Cqd1 and Cqd2 in yeast restores the normal distribution of yeast CoQ6 [93], indicating that other trafficking mechanisms must operate. Hence, it is likely that other CoQ6 transporters contribute to trafficking.
Distribution of CoQ6 between mitochondrial and non-mitochondrial membranes plays important functional roles in mitochondrial respiration, response to oxidative stress, and redox control. The cqd1Δ single mutant has decreased mitochondrial CoQ6 content, grows slowly in a medium containing glycerol as the sole non-fermentable carbon source, has increased non-mitochondrial CoQ6 content, and shows enhanced resistance to treatment with PUFAs [93]. Conversely, the cqd2Δ single mutant has increased mitochondrial CoQ6 content, normal growth on a medium containing glycerol, and is more sensitive to treatment with PUFAs [93]. The trafficking of CoQ to non-mitochondrial membranes (including the plasma membrane) mediates resistance to ferroptosis, a type of cell death due to lipid peroxidation [32,33]. CoQ6 in the plasma membrane functions as a potent antioxidant and co-antioxidant (Figure 1b), and is also in the PMRS (Figure 1c). In respiratory deficient cells (yeast and HL-60 cultured cells), there is an increased content of CoQ at the plasma membrane [97,98]. This recruitment of CoQ to the PMRS enables cytosolic NADH to be oxidized to NAD+, together with export of the electrons to extracellular acceptors, such as ascorbate, and thereby maintains the ratio of cytosolic NAD+/NADH [99,100]. Hence, the pathways that control the trafficking of both endogenous and exogenous CoQ exert profound effects on metabolism and stress response.
3. Mammalian Cell Culture Models
Mammalian cell culture has been integral in the study of the uptake and distribution of CoQ for many decades. Among the reasons for using cell culture(s) to study CoQ uptake and trafficking are the high degree of specificity with which one can control their environment, the ease and accuracy of administration of nutrients, hormones, and in this case CoQ, the decreased physiological variability as compared to live animals. However, whether cells in culture accurately reflect their true physiological context is often difficult to determine.
In addition to uptake and trafficking of exogenous CoQ, cell culture has been used to study the CoQ synthome and endogenous CoQ biosynthesis [101]. The functional roles of CoQH2 as an antioxidant have also been characterized in culture [102,103], with a specific emphasis on plasma membrane-localized CoQ as a protectant against ferroptosis [32,33,104,105]. Cells in culture have likewise proven to be a powerful model for the study of CoQ uptake and distribution. In the following section, we discuss CoQ uptake and intracellular trafficking in epithelial, endothelial, blood, dermal, and HeLa cells. Subsequently, the supplementation and efficacy of CoQ10H2 and CoQ10 will be compared, before concluding with a discussion about cell receptors that mediate uptake of exogenous CoQ10.
3.1. Studies of CoQ Uptake in Mammalian Cell Culture
3.1.1. Epithelial Cell Lines
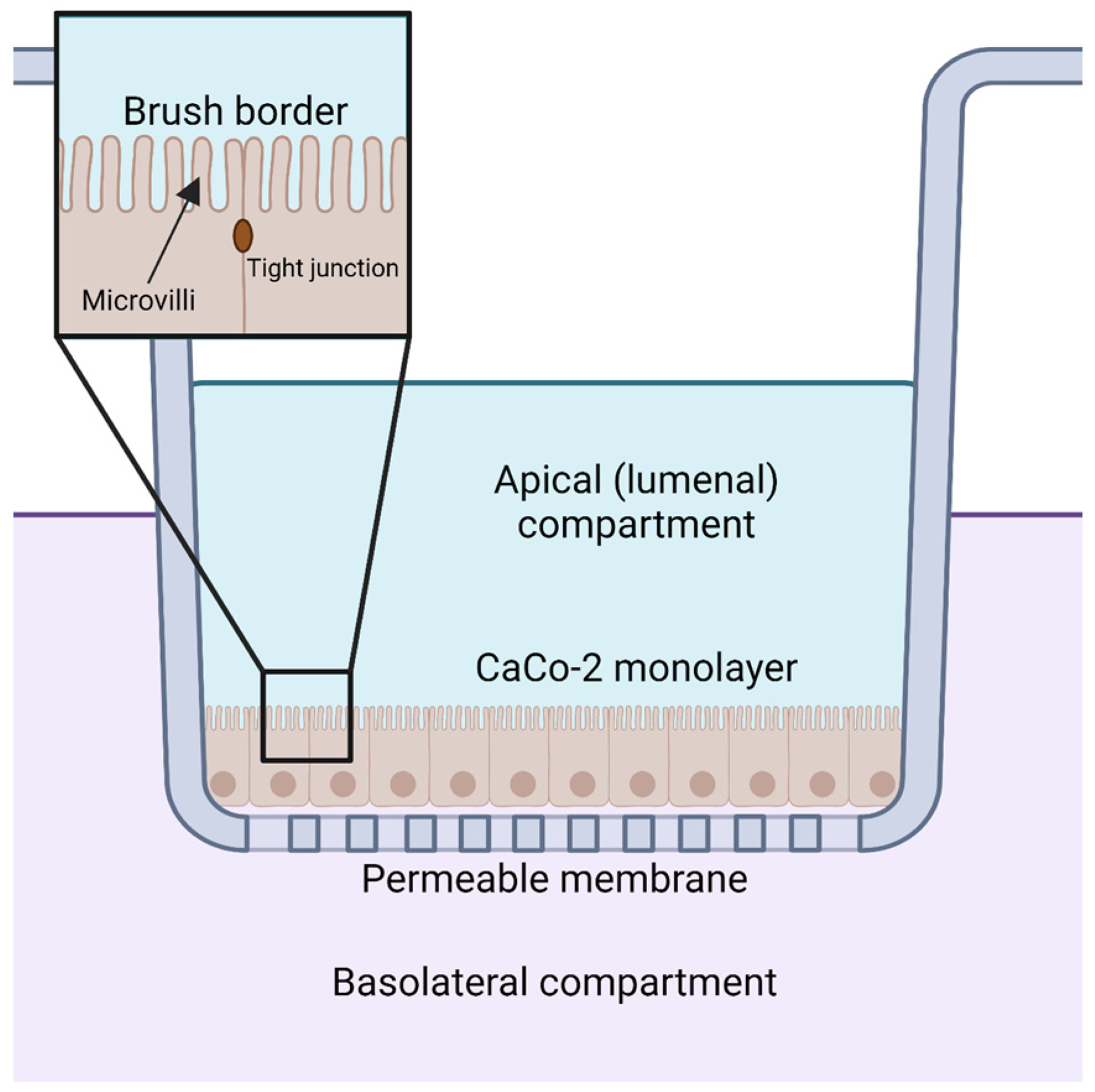
Orally supplemented CoQ10 is first taken up by the epithelial cells of the digestive tract [2,9,106]. However, the mechanism by which enterocytes absorb CoQ10 is not well understood. Several reviews and studies state that the uptake of CoQ10 occurs by a simple process of passive diffusion [2,9]. A combination of passive diffusion and receptor-mediated uptake is responsible for absorption of fatty acids [107], vitamin E [108], and cholesterol [109] in enterocytes. Niemann–Pick C1-like 1 (NPC1L1) plays an important role in uptake of dietary cholesterol; its disruption in mice results in a 50% decreased cholesterol absorption [109]. Recent studies suggest that membrane protein transporters are also important for the uptake of CoQ10. The human intestinal epithelial cell line CaCo-2 provides a model for much of the recent research on CoQ absorption in the digestive tract. CaCo-2 is a colorectal adenocarcinoma cell line that is used to study transport across the intestinal epithelium, including the absorption of various hormones and small molecules such as insulin [110], anthocyanin [111], and calcitonin [112]. Among the advantages of this cell line is its formation of tight junctions, microvilli that form a brush border on the luminal or apical side, and transport proteins that allow the uptake and digestion or transport of nutrients to the basolateral side, all of which can simulate the in vivo digestion of molecules via oral supplementation (Figure 5) [113].
CaCo-2 cells can assimilate solubilized CoQ10 from digested oral supplements, such as CoQ10 in liposomes and micelles. Supplementation of CaCo-2 cells with CoQ10-infused liposomes altered the mRNA levels of 694 genes involved in signaling, metabolism, and general transport [106]. Many of these proteins are directly regulated by CoQ, such as Caspase-3, which is inhibited by CoQ10 at the plasma membrane [114]. CoQ10 uptake by CaCo-2 cells is enhanced when solubilized forms of CoQ10 were administered via simulated digestive micellization [115]. Eight different commercial CoQ10 products were mixed with fat-free yogurt and olive oil, and subjected to gastric conditions by treatment with hydrochloric acid and porcine pepsin. Following this treatment, lipase, bile extract, and porcine pancreatin were added to simulate digestion. This digestion micellized the CoQ10, which was then extracted from the aqueous layer of the resultant mixture. Of the eight CoQ10 products, four exhibited greater than 60% micellization in the aqueous layer: γ-Cyclodextrin complexed-CoQ10 tablet, liposomal CoQ10 solution, solubilized CoQ10 capsule, and a γ-Cyclodextrin/solubilized CoQ10 powder termed Hydro-Q-Sorb® [115]. γ-Cyclodextrin is a water-soluble octasaccharide of particular interest to the optimization of CoQ uptake [116]. The sugar features eight glucose subunits interlinked to form a cylindrical shape, characterized by its hydrophilic exterior and hydrophobic interior, which can encapsulate CoQ10. γ-Cyclodextrin is one of many formulations of CoQ10 that have been assessed for their ability to enhance its bioavailability [117]. The liposomal CoQ10 and solubilized CoQ10 displayed the greatest rate of micellization, but it was Hydro-Q-Sorb® that was absorbed most by CaCo-2 cells. Hydro-Q-Sorb® exhibited a dramatic increase in CoQ10 content after a four-hour incubation when compared to incubation with the non-solubilized CoQ10 powder reference, despite Hydro-Q-Sorb® containing only 20% of the concentration of CoQ10 as the reference powder [115]. The correlation between CoQ10 product micellization and CaCo-2 uptake was weak, suggesting that other factors regulate CoQ10 uptake besides micellar transport [115]. Regardless, there does exist a positive relation between the micellization of CoQ10 and its internalization in the CaCo-2 cell model, indicating the importance of lipid dispersion in CoQ uptake.
The CaCo-2 human intestinal epithelial cell model has been used to compare the relative effects of CoQ10 and CoQ10H2 uptake, as several studies postulate that CoQ10H2 is more bioavailable than CoQ10 [118]. Supplementation with CoQ10H2 poses a challenge because the hydroquinone undergoes rapid autoxidation under cell culturing conditions [119]. As such, studies designed to compare treatment with CoQ10H2 versus CoQ10 should monitor autoxidation of the CoQ10H2 during incubation conditions. Recently, Naguib et al. [120] found that CoQ10H2 complexed with γ-cyclodextrin remained stable in an aqueous salt solution for up to seven days, with the majority of the original CoQ10H2 not undergoing autoxidation to CoQ10, demonstrating that CoQ10H2 can be supplemented with high purity in this form. Failla et al. [118] investigated the relative uptake and transport of CoQ10 and CoQ10H2 across the CaCo-2 monolayer using a previously described simulated digestion method [115]. CoQ10 or CoQ10H2 was subjected to “digestion” and added to the apical medium. Following a four-hour incubation, the total cell-associated CoQ10 content and the total amount of CoQ10 transported into the basolateral medium were determined. Both apical uptake into CaCo-2 cells, and transepithelial transport was significantly higher for the micellized CoQ10H2 as compared to the micellized CoQ10 [118]. The authors showed that incorporation of CoQ10H2 into micelles during the digestion process was twice as efficient when compared with CoQ10 [118]. Non-complexed CoQ10 has been shown to have very low permeability across the CaCo-2 monolayer [118,121], indicating that in the absence of CoQ10 incorporation into micelles or liposomes, transport across the brush border is extremely inefficient. Thus, both physiological and pharmaceutical solubilization methods for CoQ10 (or CoQ10H2) are needed to enhance its absorption across the small intestine epithelium.
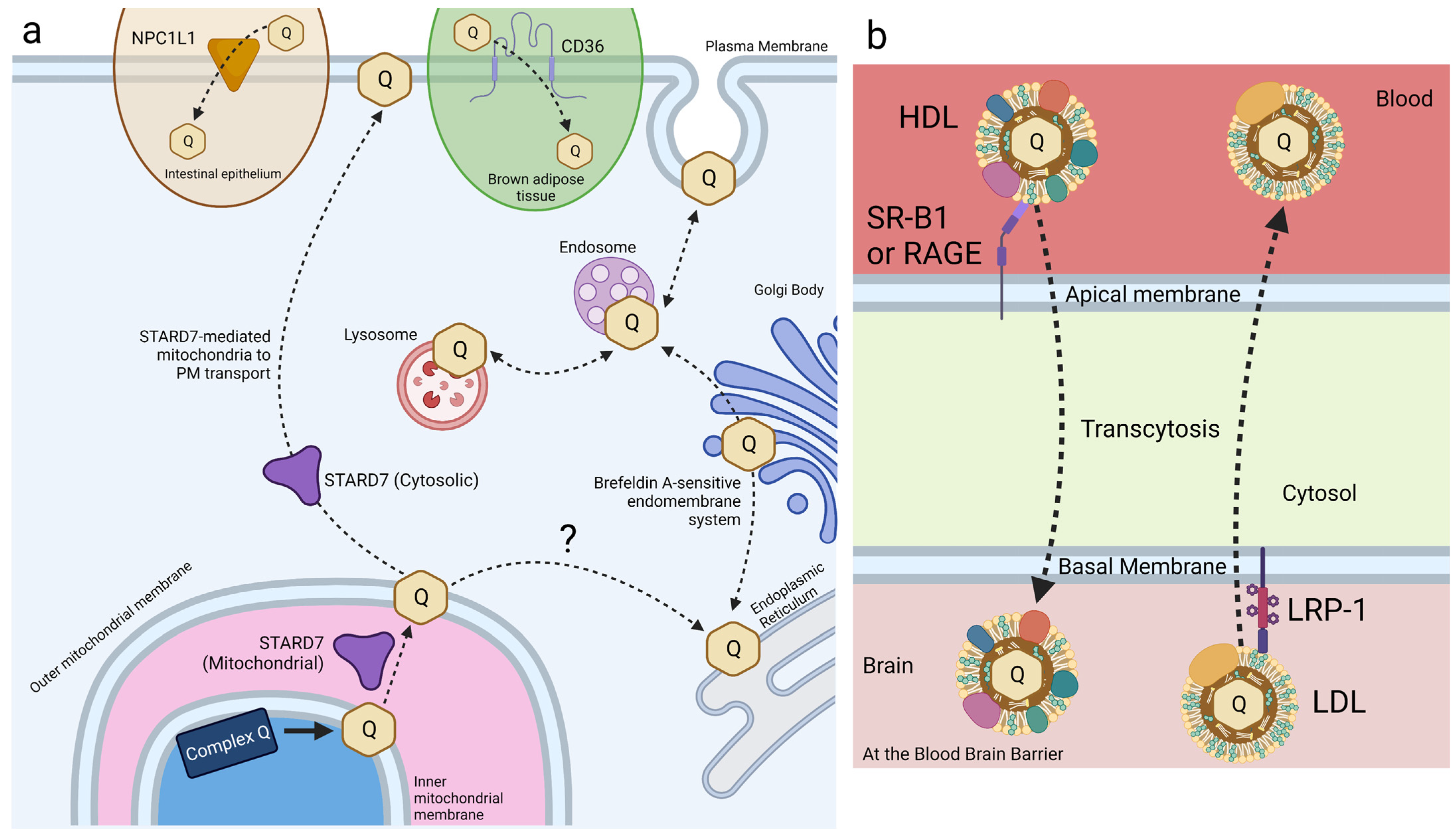
Small intestine epithelial cells express NPC1L1, a transport protein involved in cholesterol and vitamin E transport and metabolism (Figure 6a) [108,122,123]. Recent work has identified a role for NPC1L1 in the intestinal absorption of CoQ. NPC1L1 was overexpressed in Madin–Darby canine kidney (MDCK) epithelial cells to assess NPC1L1-mediated CoQ9 and CoQ10 transport [124]. MDCK cells are epithelial cells derived from kidney tissue of a cocker spaniel. NPC1L1 was expressed over 50-fold in human small intestine and duodenum compared to the kidney, making this mutant MDCK model reasonably analogous to that of intestinal CoQ uptake [125]. CoQ10 and CoQ9 uptake was found to increase significantly in NPC1L1-overexpressed MDCK cells, as compared to control MDCK cells. Furthermore, treatment with ezetimibe, a selective inhibitor of NPC1L1, decreased CoQ10 and CoQ9 uptake of NPC1L1-overexpressed cells back to control levels, implicating NPC1L1 as a CoQ-transport protein [124]. This study demonstrates the existence of intestine-specific proteins that function to increase CoQ10 absorption, among other lipids. However, the mechanism of NPC1L1-CoQ10 interaction remains unclear.
The HepG2 liver epithelial cell line has played an important role in the development of new strategies for exogenous CoQ10 uptake. Moschetti et al. [126,127] engineered a nanodisk composed of a phosphatidylcholine bilayer circumscribed by apolipoprotein-A1, where CoQ10 integrates readily within the midplane of the phosphatidylcholine bilayer. These nanodisk structures are defined as “reconstituted high-density lipoproteins” [128], and the CoQ10 is integrated into a disk-shaped bilayer. The solubilization efficiency of the nanodisk-CoQ10 was determined to be 97%, as compared to CoQ10 which is essentially insoluble when dispersed in the presence of just apo-A1 or phosphatidylcholine [126]. The authors then assessed the disk’s ability to transport CoQ10 into HepG2 cells. HepG2 cells exhibited a significant increase in mitochondrial CoQ10 content upon incubation with nanodisk-associated CoQ10 compared to empty nanodisks. The authors conclude this nanodisk to be a promising vehicle for exogenous CoQ10 uptake and distribution in liver epithelium. However, the study lacked a control where CoQ10 was supplemented in the absence of the nanodisk formation. This would have provided a useful point of comparison.
To further investigate the potential effectiveness of biomolecular nanodisks as a drug-delivery vehicle for CoQ10, cultured C2C12 myotubes were treated with supra-pharmacological doses of simvastatin, an HMG-CoA reductase inhibitor that decreases the production of polyprenyl diphosphate precursors, and nanodisk-CoQ10 [127]. Treatment with simvastatin caused a dose-dependent decrease in cell viability, reduced mitochondrial CoQ10 content, and reduced the oxygen consumption rate. However, co-treatment with simvastatin and nanodisk-CoQ10 increased mitochondrial CoQ10 content compared to simvastatin treatment alone and ameliorated the reduction in oxygen consumption rate. These results highlight the potential of nanodisks to dramatically improve the bioavailability of CoQ10. It would be interesting to assess the stability of CoQ10H2 in nanodisks and compare its uptake relative to that of the nanodisk-CoQ10. Animal studies are needed to determine the efficacy of nanodisks as a drug-delivery vehicle in vivo.
3.1.2. Endothelial Cell Lines
Following absorption of CoQ10 into the bloodstream, endothelial cells are the next point of uptake. Endothelial cells form the monolayer that line the luminal surface of blood vessels and capillaries, mediating macromolecule transport to and from neighboring tissues. The endothelium is studied extensively in the uptake and distribution of drugs due to this essential role in post-digestive tract drug transport. Human umbilical vein endothelial cells (HUVECs), human corneal endothelial cells, and bovine aortic endothelial cells (BAECs) have been utilized as models for CoQ uptake.
HUVECs have been used to study cell response to CoQ10 as a protectant against oxidative stress. Treatment of HUVECs with Amyloid-β1-42 or with a sub-fragment of Aβ1–42 (Aβ25–35) causes oxidative stress and elicits cell death via apoptosis and necrosis [129]. Pre-treatment with exogenous CoQ10 (1–7 μM) was found to reduce reactive oxygen species (ROS) generation, inhibit the uptake of Aβ25-35, and prevent cell death [130]. The results suggest that the uptake and trafficking of exogenous CoQ10 is directed to sites essential in the quenching of free radicals within HUVECs [130].
Naguib et al. recently reported the uptake of CoQ10H2 in HCEC-B4G12 human corneal endothelial cells. HCEC-B4G12 cells exhibited a higher increase in total CoQ10 content when supplemented with CoQ10H2/γ-cyclodextrin as compared to non-complexed CoQ10 [120]. Furthermore, HCEC-B4G12 cells treated with ferroptosis-inducer erastin exhibited viability akin to that of untreated cells when incubated with as little as 1 µM CoQ10H2/γ-cyclodextrin, whereas HCEC-B4G12 cells treated with erastin and 100 µM non-complexed CoQ10H2 exhibited minimal rescue [120]. Uptake was also assessed by measuring ROS production in response to antimycin A, a mitochondrial electron transport chain Complex III inhibitor [120]. HCEC-B4G12 cells demonstrated a dose-dependent rescue of ROS generation in response to supplementation with CoQ10H2/γ-cyclodextrin, whereas non-complexed CoQ10H2 produced no significant change [120]. The authors report a promising level of CoQ10H2/γ-cyclodextrin uptake by this corneal endothelial cell line, pointing towards possible exogenous CoQ10H2 formulations involving γ-cyclodextrin to protect against oxidative stress-induced loss of function in human corneas.
BAECs incubated with non-complexed CoQ10 do not exhibit significant levels of uptake. When BAECs were treated with glucose oxidase, an enzyme that oxidizes glucose to produce hydrogen peroxide, incubation with MitoQ reduced the activity of apoptotic enzymes Caspase-3 and Caspase-9, indicating rescue from oxidative stress [131]. Conversely, incubation with CoQ10 did not reduce apoptotic activity, suggesting poor uptake of CoQ10 and/or impaired trafficking to sites essential for protection against oxidative stress [131]. Furthermore, when BAECs pre-treated with 1 µM Mito-Q were incubated in either glucose oxidase or hydroperoxides, complex I activity (as measured via the rate of NADH oxidation) exhibited significant rescue compared to the untreated control; whereas pre-treatment with 1 µM CoQ10 resulted in insignificant rescue in both glucose oxidase and hydroperoxide incubated BAECs [131]. Perhaps CoQ10H2/γ-cyclodextrin treatment could improve the uptake in BAECs, like that discussed for CaCo-2 and corneal epithelial cells.
3.1.3. Human Blood Cells
The HL-60 cell line is a human myeloid leukemia cell derived from a patient with promyelocytic leukemia. These cells may be cultured in suspension. The trafficking of endogenously synthesized 14C-labeled CoQ10 was compared with the trafficking of exogenously added CoQ9 [83]. HL-60 cells were incubated with 14C-labeled 4-HB, the ring precursor of CoQ biosynthesis (Figure 2), and the content of 14C-labeled CoQ10 was determined in subcellular fractions over a period of 24 h. Fractions enriched in mitochondria contained more than 90% of the 14C-labeled CoQ10, as ascertained at time points from two to 24 h. Fractions enriched in ER and mitochondria-associated membranes (MAM) contained small amounts of 14C-labeled CoQ10 by two hours, and the plasma membrane fraction had detectable 14C-CoQ10 only after six hours [83]. HL-60 cells treated with exogenous CoQ9 showed an accumulation of CoQ9 in mitochondrial and MAM fractions, followed by its subsequent accumulation in ER and plasma membrane fractions. After twelve hours of incubation with CoQ9, the mitochondrial fraction contained the highest content of CoQ9, followed by the ER and MAM fractions with similar CoQ9 content, whereas the plasma membrane fraction exhibited significantly lower CoQ9 content [83]. The endo-lysosomal membrane fraction was most enriched in CoQ9, but this was not tracked during the time course. Treatment of HL-60 cells with brefeldin A, an inhibitor of vesicle formation and transport between the ER and Golgi, prior to the addition of 14C-4-HB resulted in a profound inhibition in the content of 14C-CoQ10 in all fractions, with the plasma membrane fraction being most decreased. It is interesting that inhibition of trafficking between the ER and Golgi had such a dramatic effect on the endogenous synthesis of CoQ10 within the mitochondria, and may indicate the importance of contact sites between the ER and mitochondria. Treatment of HL-60 cells with brefeldin A and CoQ9 resulted in most of the CoQ9 being sequestered in the endo-lysosomal fraction [83]. The high CoQ9 content in the endo-lysosomal fraction suggests the importance of the endo-lysosomal trafficking pathway in the uptake and assimilation of exogenous CoQ9. These findings indicate the importance of a functional endomembrane system in the synthesis of endogenous CoQ10 and in the transport of both exogenous CoQ9 and endogenous CoQ10.
A recent paper by Wani et al. [132] found exogenous CoQ10 (non-complexed) to ameliorate oxidative stress in erythrocytes and lymphocytes, suggesting its uptake. Erythrocytes and lymphocytes isolated from donor blood samples were exposed to TiO2 nanoparticles to induce oxidative stress. Upon treatment with TiO2, erythrocytes exhibited impaired ATPase activity and increased ROS production, and lymphocytes had severely depleted ATP levels and reduced mitochondrial membrane potential. Incubation with TiO2 in the presence of CoQ10 produced significant rescue of all markers of oxidative stress to wild-type or near-wild-type levels in both erythrocytes and lymphocytes [132]. These data indicate the ability of lymphocytes to take up and distribute exogenous CoQ10 to the mitochondria, where it restored mitochondrial membrane potential, ATPase activity, and ATP levels. Lymphocytes are also able to traffic exogenous CoQ10 to other cellular membranes, where it can slow lipid peroxidation induced by oxidative stress.
3.1.4. Human Dermal Cells
Human dermal fibroblasts (HDFs) are specialized cells found in the dermis that play a vital role in the maintenance and repair of the skin. The use of HDFs is especially relevant to investigate the efficacy of CoQ10 as a commercially available skincare and anti-wrinkle product. HDFs exhibit significant levels of exogenous CoQ10 uptake, as measured by a reduction in ROS generation [133]. The authors used pollutant particulate matter to activate KU812 cells, a basophilic leukocyte cell line, and used the resulting conditioned medium as a stress treatment of HDFs. Such treatment elicits a proinflammatory response and ROS generation in HDFs. Following eight hours of treatment, the HDFs were then treated with 100 nM CoQ10. The exogenous CoQ10 affected a significant reduction in mitochondrial ROS generation [133]. These findings suggest the ability of HDFs to take up and traffic CoQ10 to the mitochondria.
HDFs have also been used to demonstrate an efficacious uptake of liposomal CoQ10. CoQ10 was suspended into liposomes via thin-film hydration, with a drug entrapment efficiency of 73.1% [134]. Administration of this liposomal-CoQ10 suspension to HDFs that were oxidatively stressed from hydrogen peroxide treatment resulted in a complete rescue of cell viability to the point of cell proliferation, whereas non-complexed CoQ10 administration of the same concentration caused no significant change in cell viability [134]. Similarly, quantification of ROS in these HDFs revealed that liposomal-CoQ10 supplementation significantly reduced ROS production, while supplementation with non-complexed CoQ10 produced no significant change [134]. The uptake of CoQ10 by HDFs is thus augmented by liposomal suspension, where the CoQ10 can restore antioxidant function.
HDFs exhibit improved uptake of exogenous CoQ10H2 versus CoQ10. Incubation of HDFs with simvastatin decreased CoQ10 levels by 29% [135]. A 24-h supplementation with 15 μg/mL CoQ10H2 caused a dose-dependent increase in total cellular CoQ10 content, up to an 87-fold increase compared to the vehicle control. In contrast, supplementation with 15 μg/mL CoQ10 resulted in just a 4.6-fold increase, a 95% reduction compared to CoQ10H2 [135]. Isolated mitochondria in HDFs treated with CoQ10H2 also exhibited a 160-fold increase in total CoQ10 content, compared to a 2.5-fold increase with CoQ10 treatment. Thus, treatment with CoQ10H2 resulted in enhanced mitochondrial CoQ10 content relative to treatment with CoQ10 [135]. The administration of CoQ10H2 to enhance uptake is promising, but the mechanisms that produce such high uptake relative to CoQ10 remain uncharacterized.
Keratinocytes are epidermal skin cells that constitute the majority of the human epidermis, essential in forming the primary defense against environmental damage. CoQ10 treatment has recently been shown to protect keratinocytes from oxidative stress-induced cell death, indicating its uptake [136]. HaCaT human keratinocytes derived from the foreskin were incubated with CoQ10 directly added to Dulbecco’s modified Eagle’s Medium (DMEM) and fetal bovine serum (FBS), prior to treatment with the free-radical generating compound 2,2′-azobis-(2-amidinopropane) dihydrochloride (AAPH). Interestingly, no rescue of cell viability was observed until the DMEM-CoQ10 medium was enhanced with CoQ10 premixed in FBS, which resulted in a significant rescue of cell viability in the keratinocytes [136]. Similarly, intracellular CoQ10H2 content increased significantly after incubation with the premixed CoQ10 medium, as compared to both the control and the original, insolubilized CoQ10 medium [136]. The authors suggest that uptake of CoQ10 by keratinocytes is augmented by the administration of CoQ10 in a premixed medium, where otherwise an inefficient uptake is observed.
Keratinocytes have proved useful in the characterization of a new iodine-labeled CoQ derivative that may allow monitoring of the biodistribution of exogenous CoQ [137]. The novel iodine-labeled CoQ10 (I2-Q10) consists of CoQ10 with two iodine atoms replacing the terminal carbon of the polyisoprenyl tail. Iodine fluoresces upon excitation via X-rays, allowing for quantification and localization of I2-Q10 with minimal overlap between other molecules. Human keratinocytes incubated in 50 µM I2-Q10 showed no signs of toxicity or cell death, with levels of CoQ10 uptake comparable to that of unlabeled CoQ10. X-ray fluorescence imaging of pelleted keratinocytes incubated in I2-Q10 revealed a cell-specific localization of the iodine fluorescence with no extra-pellet iodine leakage. Furthermore, incubation with 50 µM I2 resulted in a significant decrease in the pellet’s fluorescence, demonstrating poor uptake of elemental iodine and thus a mechanism for I2-Q10 uptake that is independent of iodine labeling uptake per se. The authors found exogenous I2-Q10 to distribute homogeneously over the keratinocyte cell, but co-localization with subcellular organelles, such as mitochondria, was not determined. Beyond the confirmation that CoQ10 is taken up by human keratinocytes, these data suggest a potential way to assess CoQ10 distribution using I2-Q10 in X-ray fluorescence imaging [137]. The technique was demonstrated to allow for quantitative analysis with no phenotypic changes due to the iodine labeling, and it shows promise in the investigation of mechanisms underlying exogenous CoQ uptake. It is also worth investigating whether the supplementation of with I2-Q10 restores respiratory function in a CoQ-deficient cell culture model, as some CoQ derivatives suffer from a loss of function upon modification of the tail (e.g., Mito-Q).
3.1.5. HeLa Cells
HeLa cells, derived from cervical cancer cells, are the oldest and most generally used human cell line in medicine to date. Recent work in HeLa cells has identified STARD7 as a phosphatidyl transfer protein involved in the export of endogenous CoQ10 from the mitochondria to the plasma membrane, where it plays a role in ferroptosis suppression. STARD7 is localized to both the mitochondrial intermembrane space and the cytosol (Figure 6a). Deletion of STARD7 causes a nearly 5-fold increase in ferroptosis in response to treatment with PUFAs and erastin [63]. STARD7 is postulated to deliver mitochondrial CoQ10 to the plasma membrane, where it is reduced by FSP1 to CoQ1010H2 and suppresses ferroptosis. Expression of a cytosol-localized form of STARD7 in HeLa cells significantly decreased cell death in response to treatment with erastin and PUFAs. Inhibition of either CoQ10 biosynthesis or FSP1 function completely abolished resistance to ferroptosis, resulting in wildtype-levels of cell death in response to oxidative stress [63]. Expression of the mitochondria-localized form of STARD7 alone resulted in a significant decrease in plasma membrane-associated CoQ10, while the mitochondrial CoQ10 content was unaffected. The introduction of cytosol-localized STARD7 rescued plasma membrane-associated CoQ10 content, suggesting that STARD7 functions as a CoQ10 transporter in both the mitochondria and cytosol [63].
HeLa cells have also been used to study the effect of supplemented CoQ10 on sulfide oxidation. SQR is the initial enzyme in the sulfide oxidation pathway, whose impairment can lead to increased ROS. CoQ10 is an essential cofactor for SQR and protects the cell against oxidative stress. Silencing of COQ8A RNA in HeLa cells decreased the CoQ10 content by approximately 50% and led to enhanced ROS production. Supplementation of CoQ10 via Hydro-Q-Sorb decreased ROS production to near-control levels [138]. However, when SQR was knocked down in these CoQ10-deficient cells, exogenous CoQ10 no longer decreased ROS production [138]. This finding indicates that the supplementation of CoQ10 rescued ROS levels due to CoQ10-SQR activity, which requires CoQ10 localization to the inner mitochondrial membrane. Thus, HeLa cells could take up the Hydro-Q-Sorb CoQ10 and traffic it to the inner mitochondrial membrane, where it rescued sulfide oxidation.
HeLa cells have been utilized in the characterization of a lipopeptide-based surfactant, caspofungin (CF), that has been shown to solubilize CoQ. CF can form micellized nanoparticles with CoQ10 up to 200 nm in diameter, with a critical micelle concentration of 50 µM [139]. Wang and Hekimi recently found that CF-solubilized CoQ10 is a highly efficient formulation for exogenous supplementation in HeLa cells. Upon incubation for one hour, the CoQ10 content in human HeLa cells increased significantly when treated with CoQ10-CF as compared to CoQ10, demonstrating the complex’s rapid uptake. Recent results further demonstrated that treatment with CoQ10-CF restored maximal respiration in CoQ10-deficient human fibroblasts derived from a patient harboring a homozygous mutation in the COQ7 gene [140]. The mechanism for CF micellization of CoQ10 remains unknown, and the potential for CoQ10-CF as a therapeutic to treat CoQ10 deficiencies in patients warrants investigation.
3.1.6. Mouse Embryonic Fibroblasts (MEFs)
Wang et al. found that treatment with CoQ10-CF can increase the uptake of exogenous CoQ10 in MEFs in a dose-dependent manner. Incubation of MEFs for 72 h with 20 μM CF and 0.25 μM CoQ10 produced a near-doubling in basal and maximal oxygen consumption rate as compared mouse embryonic fibroblasts incubated in just 10 μM CF without exogenous CoQ10 [139]. After just a one-hour incubation with the CoQ10-CF complex, MEF CoQ10 content was approximately 3-fold higher when compared to MEFs incubated with free CoQ10 for 48 h, indicating the rapid uptake of the complex.
3.2. Cell Receptors for Exogenous CoQ Uptake and Transport
3.2.1. Receptor-Mediated Uptake of CoQ
Receptor-mediated endocytosis plays a key role in the selective uptake of extracellular molecules, including CoQ10. Wainwright et al. [141], used an in vitro blood–brain barrier (BBB) endothelial cell model of CoQ10 deficiency, and identified lipoprotein-associated uptake and efflux mechanisms regulating CoQ10 distribution across the BBB (Figure 6b). Porcine brain endothelial cells (PBECs) were grown in monolayer on a permeable membrane (see Figure 5), and transport of CoQ10 was assessed from both the apical to basal (A to B, blood-to-brain side), and basal to apical (B to A, brain-to-blood side). The transport of non-complexed CoQ10 across porcine brain endothelial cells (PBEC) was extremely low. Pre-treatment of CoQ10 with bovine plasma-derived serum led to its association with the VLDL/LDL lipoprotein fractions and increased CoQ10 transport across the PBEC monolayer by 4-fold, suggesting that CoQ10 may be transported across the BBB as part of a lipoprotein complex. Transport of serum-treated CoQ10 in the A to B direction matched transport in the B to A direction, indicating that opposing transport systems produced no net accumulation of CoQ10 in the brain. This was observed for both the PBEC model and for a mouse BBB cell line, bEnd.3 [141]. Transporters at the BBB generally act together to limit systemic lipoprotein and cholesterol transfer to the brain.
Based on known lipoprotein transporters at the BBB, pharmacological inhibitors were screened for their effect on CoQ10 transport across bEND.3 cells. BLT-1 is an inhibitor that targets the scavenger receptor (SR-B1), which interacts with HDL on the apical membrane to execute caveolae-mediated lipoprotein influx [142]. RAP was used to inhibit the low-density lipoprotein receptor (LDLR) family members, including the LDL receptor-related protein 1 (LRP-1). LRP-1 interacts with a variety of ligands to mediate BBB integrity [143]. FPS-ZM1 was used to inhibit the receptor for advanced glycation end products (RAGE). RAGE mediates the influx and opposes LRP-1-mediated efflux of ApoE and amyloid-beta across the BBB [144]. Upon inhibition of either SR-B1 or RAGE, apical-to-basal transport of CoQ10 was decreased to 44% and 50% of the control, respectively, whereas basal-to-apical transport exhibited no change [141]. The authors suggested a direction-specific role of both receptors for CoQ10 transcytosis into the brain. In contrast, inhibition of LRP-1 (and LDLR family members) increased apical-to-basal CoQ10 transport by 168%, with no significant change in the reverse transport, suggesting that LRP-1 directs CoQ10 transport out of the brain [141].
Wainwright et al. used pABA, a competitive inhibitor of polyprenyldiphosphate-4-hydroxybenzoate transferase (Coq2), to create a CoQ10-deficient model of the BBB. Treatment with pABA significantly decreased cellular CoQ10 content and mitochondrial respiratory chain complex activities in bEND.3 cells [141]. Cells treated with pABA also had reduced tight junction integrity and increased permeability (paracellular transport, Figure 6b), causing a shift in the net transport of CoQ10 in the blood-to-brain direction. Under these pABA-inhibited conditions, the RAGE receptor was responsible for some of the net CoQ10 transport, while the SR-B1 receptor-mediated transport was no longer active. However, the use of pABA to inhibit CoQ10 biosynthesis has possible confounding effects. pABA is a substrate for Coq2, and the resulting 4-amino-3-polyprenylbenzoic acid may exert negative effects on cell endocytic trafficking (see Section 2.1). Studies with mammalian cultured cells show that once pABA is prenylated, it is converted to a redox-active “dead-end” amino-containing analog of DMQ, which may interfere with respiratory electron transport [47]. Instead of pABA, it has been suggested that 4-nitrobenzoic acid (4-NB) should be used as a competitive inhibitor of Coq2, since it is not prenylated [47].
Interestingly, co-supplementation of CoQ10 with α-tocopherol or Trolox, a water-soluble analog of α-tocopherol that does not interact with lipoproteins, in pABA-treated and untreated bEND.3 cells caused an increase in CoQ10 efflux in the brain-to-blood direction. This result suggests that the co-supplementation of CoQ10 with α-tocopherol for individuals with CoQ10 deficiency might decrease CoQ10 delivery to the brain [141]. More work is needed to determine whether this effect is due to excessive antioxidants blocking uptake of lipoproteins containing CoQ10 or if mechanisms of CoQ10 uptake are mediated by changes in oxidative stress.
Finally, ABCB1 (P-glycoprotein) was studied since it is reported to decrease CoQ10 transport across the CaCo-2 intestinal epithelial-barrier model [145]. Inhibition of P-glycoprotein with verapamil had no effect on CoQ10 transport across the BBB, implying a mechanistic difference between exogenous CoQ10 uptake in small intestinal epithelial cells versus BBB endothelial cells [141]. However, Wainwright et al. note that in the experiments by Itagaki et al. [145] with CaCo-2, exogenous CoQ10 was supplemented in its free, un-micellized form, which is not favorable for small intestine uptake CoQ10. The existence of brain endothelial membrane receptors that enhance CoQ10 transcytosis in a selective direction across the BBB opens the possibility for antagonistic and/or agonistic supplements to increase exogenous CoQ10 delivery to the brain.
CD36 is another receptor that has been identified in the uptake of exogenous CoQ. CD36 is a member of the SR family of transmembrane glycoproteins and a class-B scavenger receptor that exhibits high structural and functional homology to SR-B1 [146]. CD36 transports fatty acids and plays a regulatory role in lipid metabolism [147]. HEK293 human embryonic kidney cells overexpressing CD36 took up 57% more CoQ9 than wild-type cells [148]. Importantly, receptor-mediated uptake of exogenous CoQ9 and CoQ10 was observed only when solubilized with Intralipid®, an emulsion of soybean oil, glycerol, and egg yolk phospholipids. Receptor-mediated uptake was not reported for solvent- or detergent-solubilized CoQ [148]. Additional work by Anderson et al. [148] reports that CD36 drives CoQ uptake in brown adipose tissue (BAT) and is required for normal BAT function (see Section 4.1).
3.2.2. Saposin B Binds CoQ10 and Regulates CoQ10 Content
Prosaposin is a precursor protein that undergoes proteolytic cleavage to generate four small proteins known as saposins (saposins A, B, C, and D). Saposin B is a heat-stable glycoprotein that is detected in many tissues. It forms shell-like dimers to bind sphingolipids and glycerol-phospholipids, and primarily localizes to lysosomes [149,150,151]. CoQ10 bound to saposin B was first identified in protein fractions collected from human urine samples and later detected in cell-lysates from HepG2 hepatocytes and human sperm [65]. CoQ10 was shown to be transferred to erythrocyte ghost membranes when incubated with saposin B-CoQ10 complexes at pH 4.5, suggesting that saposin B may play a role in delivery of CoQ10 to acidic compartments, such as the lysosome [65].
Saposin B has high binding affinity for CoQ10 as well as CoQ9 and CoQ7 isoforms, but a low affinity for α-tocopherol at pH 7.4 [65]. His-tagged saposin B expressed in E. coli was also shown to bind CoQ10 in in vitro experiments in a pH-dependent manner, and showed that glycosylation of saposin B is not essential for CoQ10 binding [152]. Saposin B has a comparable affinity for γ-tocopherol and CoQ10 at neutral and basic conditions, but a higher affinity for γ-tocopherol at acidic conditions, suggesting that the binding affinity of saposin B for γ-tocopherol is greater than that of CoQ10 [64]. In binding competition assays, two lipids in hexanes were mixed with aqueous saposin B at pH 7.4 and the resulting aqueous saposin B-bound lipids were measured. Interestingly, saposin B bound γ-tocopherol over CoQ10 despite similar binding affinities and bound α-tocopherol over CoQ10 despite lower binding affinity [64]. Based on known interactions of saposin B dimers with phospholipids, the authors postulated that saposin B dimers could bind two molecules of CoQ10 or one molecule of CoQ10 and two molecules of tocopherol. Their results suggest that the binding of one molecule of CoQ10 may accelerate the binding of two molecules of α-tocopherol. The crystal structure of saposin B binding chloroquine was later resolved [153] and molecular docking studies identified two binding sites that are accessible to a variety of ligands [154].
Mutations in prosaposin and changes to its cellular content have been reported to affect CoQ10 levels and mitochondrial function. Knockdown of prosaposin or overexpression of a prosaposin mutant that cannot form saposin B in HepG2 cells causes a significant reduction in both whole-cell and mitochondrial CoQ10 content [155]. Likewise, overexpression of prosaposin leads to an increase in whole-cell and mitochondrial CoQ10 content, suggesting that prosaposin and/or saposin B plays a role in regulating CoQ10 levels [155]. Knockdown of prosaposin in CaCo-2 cells also decreases cellular CoQ10 content, causing a decrease in ATP formation and glucose consumption [156]. As observed previously, a decrease in ATP formation is associated with disruption in tight junction formation and increased permeability.
More recent work has modeled long-term CoQ10 deficiency in HepG2 cells and found that decreased CoQ10 content was associated with decreased prosaposin levels [157]. Treatment with 4-NB decreased CoQ10 cellular content after three days and decreased prosaposin protein levels after three months. Decreased CoQ10 content and prosaposin protein levels were observed up to sixteen months with 4-NB treatment. The addition of the CoQ precursor 4-HB to cells treated with 4-NB significantly rescued both CoQ10 content and prosaposin protein levels. Studies in prosaposin-deficient mice have not investigated CoQ content, though these mice often die one to two days after birth [158]. In summary, the disruption of prosaposin or saposin B decreases CoQ10 content, indicating that saposin B is a CoQ10 transporter that plays a role in regulating CoQ10 levels.
4. Rodent Models
Mus musculus (mice), Rattus norvegicus (rats), and Cavia porcellus (guinea pigs) are common rodent models for the study of CoQ biosynthesis and uptake. Both mice and rats endogenously synthesize CoQ9 as the predominant CoQ isoform, with CoQ10 present as a minor isoform. This is in contrast to humans, guinea pigs, and other mammalian models, where CoQ10 is the major isoform [8]. Rodent models are also used to study the effect of genetic mutations on endogenous CoQ biosynthesis [159]. Hence, rodent models have been used to assess how CoQ supplements affect the distribution and content of CoQ in tissues in both normal animals as well as those harboring gene mutations causing primary CoQ deficiency [6,160].
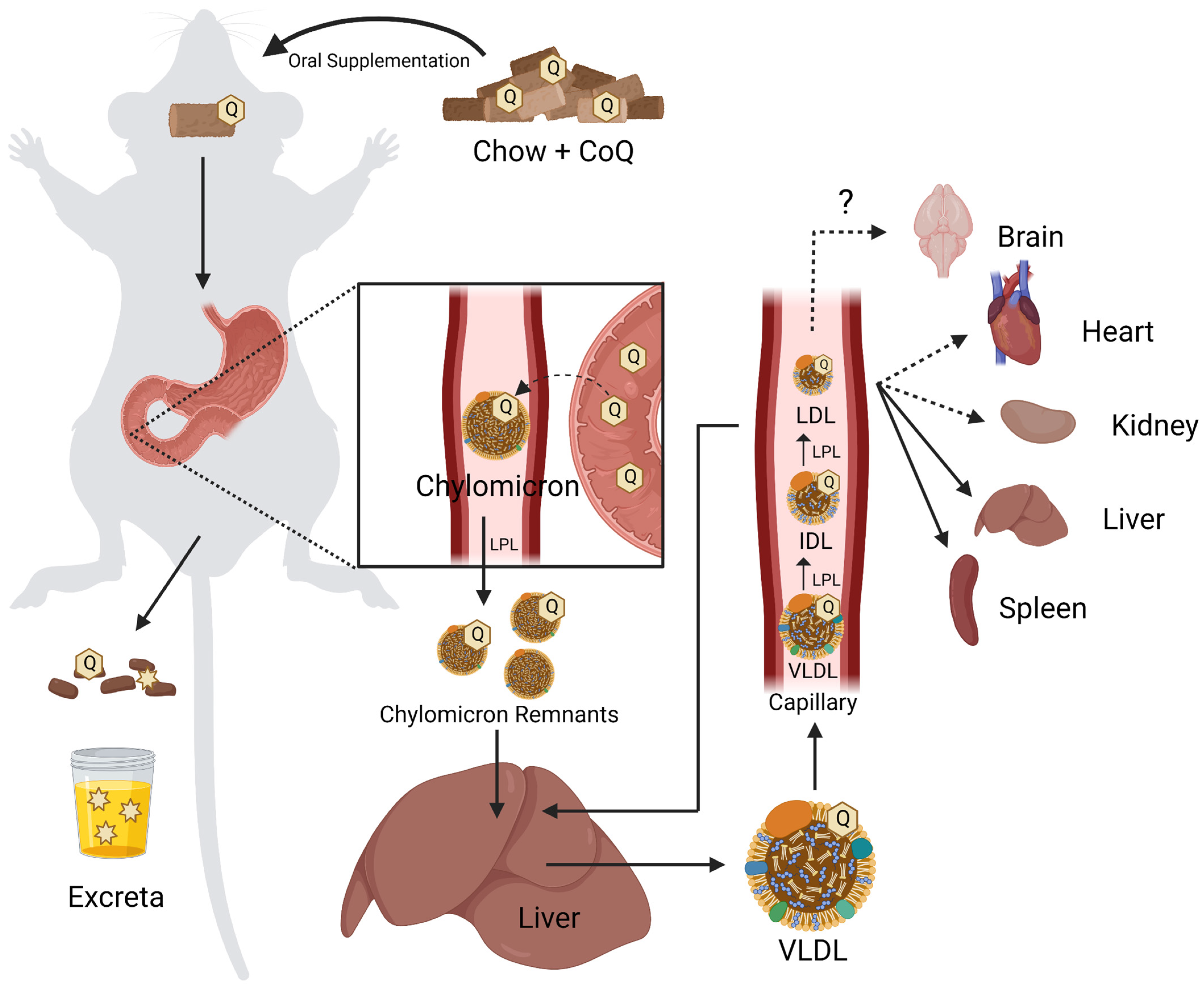
4.1. Organism-Level Uptake of CoQ
Due to their insolubility in water CoQ9 and CoQ10 are poorly bioavailable, necessitating unusually high doses [2,7]. Dosing in humans averages around 200 mg of CoQ10 taken twice daily. However, when used to treat neurodegenerative diseases, doses can reach up to 3000 mg per day [161]. As a high-molecular-weight, lipophilic molecule, CoQ10 is poorly absorbed by the gastrointestinal (GI) tract [162]. However, the consumption of CoQ10 with other dietary lipids, as well as various supplement formulations, can enhance its uptake [7,162].
A majority of rodent studies investigate the effects of CoQ10 supplements via oral treatment. The addition of CoQ10 to rodent chow is a common feeding method. Similar to studies in human patients, high doses have been used to study CoQ10 uptake in mice and rats [162,163]. As the CoQ10-supplemented chow is digested, the hydrophobic CoQ10 is taken up at the GI tract. In male Sprague Dawley rats, CoQ10 permeability along the GI tract was investigated using a side-by-side diffusion apparatus, in which isolated GI tract tissues were placed in between a CoQ10-containing donor medium and a receptor chamber [164]. These studies indicated that the greatest permeability is located at the duodenum, followed by the colon and the ileum.
Within the intestinal enterocytes, absorbed CoQ10 is likely to be packaged along with other dietary lipids into chylomicrons, which are transported into the lymphatic system [165]. Chylomicrons in the lymph are then delivered to systemic circulation via the thoracic duct and subjected to processing by lipoprotein lipase in the capillaries. The resulting chylomicron remnants are taken up by the liver (Figure 7) [166]. This delivery may be responsible for CoQ10 enrichment seen in the liver. We note here the assumption that orally supplemented CoQ10 is trafficked in a manner similar to other dietary lipids such as vitamin E [167] and cholesterol [168]. However, there is a lack of direct experimental support for the presence of supplemented CoQ10 in chylomicron remnants.
In the liver, CoQ10 derived from dietary sources is likely to be combined with de novo synthesized CoQ where it would be assembled into lipoprotein particles, with the majority in very low-density lipoprotein (VLDL) [164]. VLDL secreted by the liver is processed by lipoprotein lipase in the capillaries of various tissues to form intermediate-density lipoprotein and low-density lipoprotein (LDL) [168]. Lipoprotein particles extracted from human plasma find LDL to be most enriched in CoQ10 content [169]. CoQ10H2 is an important antioxidant in circulating lipoproteins and enhances the resistance of LDL to lipid oxidation [170,171].
High-density lipoprotein particles (HDL) isolated from human plasma contain approximately 50% of the CoQ10 content as compared to LDL [169]. This relationship between LDL and HDL CoQ content is also observed in mice [148]. In a mouse model, the absence of apolipoprotein ApoA1, a major component of HDL, decreased mitochondrial cardiac CoQ content by 75% [172]. Following ischemia/reperfusion of the left ascending coronary artery, the infarct size increased significantly in the homozygous ApoA1−/− mice, as compared to the heterozygous ApoA1−/+ mice, which in turn had larger infarcts than the parental control (WT) mice [172]. Although oral supplementation of CoQ10 in ApoA1−/− mice did not affect the cardiac CoQ10 content, intraperitoneal (IP) injection of CoQ10 restored the cardiac mitochondrial CoQ10 content of the ApoA1−/− mice to WT levels, and decreased their infarct size to that of WT mice following ischemia-reperfusion [172]. These results suggest a possible role for ApoA1 in the uptake and trafficking of CoQ10 from the GI tract to the heart; however, further study is needed to confirm this relationship. It is unfortunate that in this study the authors did not quantify the endogenous CoQ9 content as it may have further informed the outcome of supplementation with CoQ10. However, another confounding factor is that the authors used CoQ9 as an internal standard for the quantification of CoQ10. This is problematic for two reasons: (1) mouse heart contains endogenous CoQ9, and (2) commercially available standards of CoQ9 can be contaminated with CoQ10.
It is important to note that many studies in mice and rats utilize CoQ10 as the supplemented isoform, although the predominant endogenous isoform is CoQ9. This may generate conflicting outcomes when comparing rats or mice with guinea pigs, which solely produce CoQ10 as the endogenous isoform. Several studies in both rats and mice have shown that supplementation of CoQ10 increases endogenous CoQ9 levels in various tissues, serum, and mitochondria [163,173,174]. This is likely due to a sparing effect of supplemented CoQ10 on the endogenous CoQ9 pool in which the addition of CoQ10 to the cellular CoQ pool “spares” the turnover of CoQ9; a similar phenomenon is observed when CoQ10 supplements preserve levels of α-tocopherol or the tissue content of CoQ9 [175,176]. The cardioprotective effects of CoQ9 and CoQ10 supplementation were investigated in guinea pigs [177]. CoQ9 and CoQ10 were found to have functionally identical effects, as both isoforms conferred comparable cardioprotection “as evidenced from the comparable degree of the postischemic ventricular recovery and reduction in myocardial infarct size and cardiomyocyte apoptosis”. Lekli et al. further claimed that CoQ9 can be converted to CoQ10 after supplementation, although no metabolic labeling of the fed CoQ9 was performed to enable a more substantive conclusion. It is more likely the elevation in CoQ10 content was due to the aforementioned sparing effect.
CoQ10 present in lipoproteins may be delivered to cells via receptor-mediated uptake, to facilitate the entry of CoQ at the cellular level [2]. A specific example is the CD36 scavenger receptor-mediated uptake of exogenously supplied CoQ10 into mouse brown adipose tissue (BAT) [148]. CD36 is present in many tissues, with the highest abundance in adipose tissue, heart, lung, and muscle, and less expression in spleen and liver [148]. BAT functions in non-shivering thermogenesis, a process generated by uncoupled respiration [178]. To perform this function, BAT depends on the uptake of CoQ rather than through its own de novo biosynthesis [148]. BAT isolated from mice lacking the CD36 receptor (CD36−/−), have significantly reduced endogenous CoQ levels. Uptake of Intralipid®-solubilized CoQ10 in CD36−/− mice via intraperitoneal injection is significantly reduced in BAT compared to wild-type mice, while no difference was observed in liver or muscle tissues. Moreover, CD36−/− mice show a significant increase in CoQ10 serum levels after intraperitoneal injection of exogenous CoQ10 when compared to wild-type mice, suggesting that BAT is a major sink for exogenous CoQ. CD36−/− BAT mitochondria showed impaired substrate utilization and decreased basal rates of respiration, which was rescued upon the addition of CoQ2. CD36 is found to be exclusively on the plasma membrane in BAT, and BAT lacking CD36 lacks the characteristic browning of BAT following cold exposure. Likewise, CD36−/− mice exhibit defective non-shivering thermogenesis. The mechanism by which CD36 mediates the uptake of CoQ9 or CoQ10 into BAT is not clear. The authors suggested that uptake could proceed either via CD36-mediated endocytosis of lipoprotein particles or that CoQ could be selectively taken up from a bound lipoprotein [148]. In either case, these findings provide evidence for receptor-mediated uptake of CoQ. (See Section 3.2.1 for further discussion of receptor-mediated uptake of CoQ10 at the blood–brain barrier.)
Extensive reviews and literature compare the efficacy of supplementation with CoQ10 and CoQ10H2 supplementation in humans [2,7,179]. However, few studies investigate the relative bioavailability of CoQ10H2 relative to CoQ10 in a rodent model. In rats, CoQ10 is converted into CoQ10H2 during or after absorption by the intestine [162], yet exogenous supplementation with CoQ10H2 has been posited to increase the molecule’s bioavailability. Mice lacking the ability to synthesize CoQ due to homozygous partial loss of function mutations in Coq9 (Coq9−/−) were supplemented with water-soluble formulations of both CoQ10 and CoQ10H2 [180]. While both formulations increased tissue levels relative to the Coq9−/− control, the tissues of mice fed CoQ10H2 tended to have greater enrichment in CoQ10 compared to mice fed CoQ10 [180]. Measurement of cerebrum mitochondrial CoQ10 content revealed the same trends. A recent study used positron emission tomography imaging to detect the uptake and distribution of 11C-CoQ10 and 11C-CoQ10H2 that were administered to rats via tail vein injection [181]. The uptake of the labeled CoQ10H2 in the cerebrum, cerebellum, white adipose tissue, muscle, kidney, testis, and BAT was higher than that of the labeled CoQ10. However, uptake of the 11C-labeled CoQ10H2 was lower in the spleen as compared to labeled CoQ10. Although the delivery of CoQ10H2 in this 11C-labeling study was via intravenous injection [181], it is interesting that the enrichment achieved by CoQ10H2 in brain tissues was also observed for diet-delivered CoQ10H2 in the Coq9−/− mouse study [180].
4.2. Tissue Localization of Supplemented CoQ
Understanding the localization of supplemented CoQ to specific tissues may be important to treating primary CoQ deficiencies that affect specific or multiple organs. However, as discussed above, the enrichment of CoQ in specific tissues of rats and mice can be affected by the redox state of the supplement. There is also variability observed in rats, mice, and guinea pigs following supplementation with different formulations of CoQ. This variability can be attributed to the conditions of supplementation including dose, mode of supplement administration, lipid extraction, and quantification methods used, as well as the genetic background or strain of the animal model.
4.2.1. Rat
IP injection into rats of 3H-CoQ10, labeled at the isoprenyl methylene closest to the benzoquinone ring, permitted quick evaluation and extensive uptake into a number of tissues. Enriched tissues include the liver, spleen, adrenals, and ovaries, with limited uptake into the heart and thymus, as well as white blood cells [182]. This is in contrast to the poor dietary uptake of 3H-CoQ10. Over a two-week period of oral supplementation, only 4–6% was taken up while the rest was excreted as radioactive metabolites in urine and feces. Analysis of the excreta revealed that half of the radioactive content excreted was present as non-metabolized CoQ10 in feces, suggesting that excess CoQ10 is removed through bile excretion (Figure 7) [182]. The results concur with prior studies that showed both oral and IP injections of CoQ10 resulted in liver and spleen enrichment, while none was detected in the kidney, heart, or brain tissue [173,183]. However, Kwong et al. [174] observed modest CoQ10 enrichment of the heart and kidney tissues. Differences in enrichment between studies may be attributed to dosage and method of feeding. Reahal and Wrigglesworth and Lonnrot et al. supplemented CoQ10 in suspension [173,183], whereas Kwong et al. appear to supplement the chow with powdered CoQ10 [174]. CoQ10 is better taken up as an emulsion compared to a powder [2,7].
Intravenous (IV) administration in rats of 4 mg/kg CoQ10 solubilized in lipid microspheres or chromophore EL has also been investigated [184]. Chromophore EL was described as a polyethoxylated castor oil that contained approximately 40 units of ethylene oxide to each triglyceride unit along with the addition of undefined antioxidants [185]. Results from Scalori et al. [184] indicate that CoQ10, whether orally or IV administered, leaves the blood plasma quickly and is particularly enriched in the liver, although with varying kinetic profiles. The heart and kidneys of these rats were also enriched in CoQ10. However, smaller doses of CoQ10 were needed to achieve the same effect when supplemented by IV [184].
Topical application of CoQ10, a lesser-studied method of administration, has also been found to increase skin concentrations in Sprague Dawley rats [184]. This is consistent with more recent data indicating that topical administration of CoQ10 on the forearms of human females significantly increased levels at the skin surface and the deeper epidermis layer [186], and reduced UVB-induced wrinkle formation [187].
4.2.2. Mouse
Similar results for tissue enrichment of CoQ are seen in mouse models. Following a 13-week regimen of CoQ10 supplementation in a soybean oil suspension, the serum and liver homogenate had increased levels of CoQ10 [175]. No changes to CoQ10 content were observed in the brain, heart, kidney, or skeletal muscle homogenates. However, a higher dose, 200 mg/kg/day, and a longer regimen of CoQ10 were shown to increase cerebral cortex levels of CoQ10, CoQ10H2, and CoQ9H2 [188]. These results are recapitulated in a study looking at the short-term administration of CoQ10H2 [189]. Imaging mass spectrometry also shows increased CoQ10H2 in several regions of the brain, including the hippocampus, cerebellum, and brain stem [189]. Matthews et al. showed oral supplementation of CoQ10 exerted neuroprotective effects by reducing striated lesion volumes produced from systematic administration of 3-nitropropionic acid, a complex II inhibitor [188].
4.2.3. Guinea Pig
In the guinea pig model, Passi et al. [190] investigated the effect of CoQ10 and ubiquinol10-diacetate (CoQ10-DIA) provided in the diet, on liver subcellular fraction content. CoQ10H2 modified by acetate at the ring 1,4 positions, (CoQ10-DIA) is readily hydrolyzed and taken up as CoQ10H2. The study found that both CoQ10 and CoQ10-DIA taken up were mainly enriched in the heavy mitochondria and light mitochondria/lysosome fractions. Modest increases in CoQ10 content were also seen in the nucleus/debris and microsome fractions. There were no significant differences in subcellular enrichment between CoQ10 and CoQ10-DIA [190].
Across all three rodent models, exogenously supplemented CoQ10 is enriched mainly in the liver, plasma, and serum. While there remains conflicting data on CoQ enrichment in other tissues, it appears that high dosage, IV, or IP administration may be able to overcome certain barriers to uptake into tissues such as the brain, heart, and kidney.
4.3. Subcellular Localization of Supplemented CoQ
When the CoQ sequestered in lipoproteins is taken up by a cell, it must be trafficked to intracellular membranes. Currently, little is known about the mechanisms of intracellular trafficking used to shuttle CoQ. However, the subcellular enrichment of CoQ following supplementation has been studied in mice, rats, and guinea pigs.
4.3.1. Rat
Under normal physiological conditions, endogenous CoQ9 in rat liver tissue is predominantly located in the mitochondrial subcellular fraction [183]. After CoQ10 supplementation, subcellular fractionation of hepatic tissue cells was investigated [191]. The pellet containing lysosomes and light mitochondria was especially enriched in CoQ10, with no change to the endogenous levels of CoQ9. Bentinger et al. [182] conducted an investigation into the subcellular localization of 3H-CoQ10 in the liver, finding that nearly 60% of radiolabeled CoQ10 localized to the lysosome after IP supplementation, whereas the mitochondrial fractions contained only approximately 11% of the total 3H-CoQ10 recovered [182]. This is consistent with a prior study, where IP administered CoQ10 was primarily localized to the soluble cytosolic fraction of liver cells and was recovered as the oxidized form, not the reduced form [183]. As CoQ10 is highly hydrophobic, it is unclear why the concentration was found to be greatest in the soluble cytosolic fraction. It is also worth mentioning that this study does not indicate precautions taken to mitigate autoxidation of reduced CoQ10H2 in liver extracts during tissue homogenization and lipid extraction. Such precautions may include quick procedural timing, the use of acidified solvents during extraction, and inert gases to prevent autoxidation. An LC–MS/MS quantification method sensitive to the CoQ redox state was recently developed to minimize measurement error due to autoxidation [119].
In a separate rat study, the brain and brain mitochondria were shown to be enriched in CoQ10 following a two-month supplementation regimen at a dose of 200 mg CoQ10/kg per day [188]. However, the CoQ9 content was not significantly increased in this study. Another study administering 150 mg/kg CoQ10 per day for 13 weeks demonstrated an increase in heart, skeletal muscle, liver, and kidney mitochondrial CoQ10 content, as well as an additional increase in CoQ9 content in mitochondria of heart, skeletal muscle, and kidney tissue homogenate [174]. Only the mitochondria-enriched fractions were examined for CoQ9 and CoQ10 contents in these two studies.
4.3.2. Mouse
Mice supplemented with CoQ10 generally presented with increases in both CoQ9 and CoQ10 content in the liver, heart, kidney, and skeletal muscle tissue mitochondria, but not in the brain [163,176]. The data obtained by Sohal et al. were subjected to further analyses to consider the length of treatment and dosage factors. General trends indicated that mice treated for 17.5 months with CoQ10 had larger magnitude increases in CoQ content as compared to mice treated for 3.5 months [163]. No consistent dose-dependent trend was reported. However, these conclusions are solely based on the examination of mitochondria-enriched fractions
4.3.3. Guinea Pig
In the guinea pig model, Passi et al. [190] investigated the effect of dietary CoQ10 and CoQ10-DIA on liver subcellular fraction content. The authors found that both CoQ10 and CoQ10-DIA taken up were mainly enriched in the heavy mitochondria and light mitochondria/lysosome fractions. Modest increases in CoQ10 content were also seen in the nucleus/debris and microsome fractions. There were no significant differences in subcellular enrichment between CoQ10 and CoQ10-DIA.
4.4. Intracellular Trafficking of CoQ
There is little information on the intracellular trafficking of exogenously supplemented or endogenously synthesized CoQ in rodent models. Data gleaned from other cellular models, including Saccharomyces cerevisiae and human cell lines, can inform such studies. Intriguingly, ADCK2, the mouse homolog of yeast Cqd1 (discussed in Section 2.3), has been shown to play a role in modulating the transport of lipids into mitochondria for fatty acid β-oxidation and CoQ biosynthesis [95]. ADCK2 and Cqd1 are members of the UbiB/Coq8 family of atypical kinases. Adck2+/− mice exhibit haploinsufficiency, with CoQ deficiency specifically in skeletal muscle and defects in mitochondrial fatty acid oxidation [95]. CoQ10 supplementation increased the content of CoQ10 in the skeletal muscle of the Adck2+/− mice, enhanced physical performance, and decreased plasma lactate levels [95]. It seems likely that ADCK2 acts similarly to Cqd1, the yeast ortholog, and is necessary to retain CoQ within mitochondria [93].
4.5. Mouse Models of CoQ Deficiency and the Effects of CoQ Supplementation
Investigation of human mutations in COQ genes has been studied in transgenic mouse models to assess the organism-level effect of canonical single nucleotide variants. Transgenic mice with homologous gene KOs have also been used to assess and investigate phenotypic consequences, and to evaluate the efficacy of treatment with CoQ10 supplements.
Saiki et al. [192] investigated the efficacy of CoQ10 supplementation in the Pdss2kd/kd (kidney disease) mouse. The Pdss2kd/kd mouse harbors a spontaneous homozygous V117M mutation resulting in a partial loss of function in PDSS2, one of the two subunits of polyprenyldiphosphate synthase essential for CoQ biosynthesis, and a homolog of yeast Coq1 [192,193]. Pdss2kd/kd mice present with CoQ deficiency in early adulthood, resulting in a kidney disease similar to the collapsing glomerulopathy variant of focal segmental glomerulosclerosis [192]. Mice harboring a podocyte-specific loss of function knockout of Pdss2 reproduced a more severe phenotype of the Pdss2kd/kd mice, indicating that the kidney disease in the Pdss2kd/kd mouse results from the CoQ deficiency in the podocytes [193]. Intriguingly, supplementation with CoQ10 afforded a dramatic rescue of the kidney disease as ascertained by a 5-fold reduction in urine albumin content. However, CoQ10 supplementation, even at doses of 200 mg/kg per day did not affect the kidney CoQ10 content as compared to the untreated Pdss2kd/kd mouse. It was speculated that the CoQ10 supplementation may exert its effects via the filtration of blood plasma components [194]. The podocyte foot processes act as a filtration barrier, and the CoQ10 may function primarily as a membrane antioxidant [192]. A more recent study using the same Pdss2kd/kd mouse model supports CoQ10 supplementation (0.5% CoQ10 supplemented food), as a preventative treatment for the progression of nephrotic syndrome induced by deficiency [138]. Kidney CoQ9 and CoQ10 levels were increased, and damage indicated by renal histology is significantly dampened after six months of supplementation with CoQ10. CoQ10 supplementation rescued defects in the oxidation of sulfide, and decreased oxidative stress in the kidneys of Pdss2kd/kd mice [138].
The kidneys of Pdss2kd/kd mice are shown to have a defect in the sulfide oxidation pathway resulting in increased hydrogen sulfide and depleted glutathione [195]. Deficiencies in CoQ impede the sulfide oxidation pathway by decreasing SQR activity [195,196]. Further investigation of mouse Coq9 mutants revealed similar results. Coq9R239X mice, with 10-15% residual CoQ9 content, had significantly less SQR levels in the kidneys and cerebrum whereas Coq9Q95X, with 40–50% residual CoQ9 content in the same tissues, had no decrease in these tissues [196]. Both models exhibit 10–20% residual CoQ9 content in muscle and have decreased SQR in this tissue. While low CoQ content disrupts sulfide oxidation, supplementation with 100 μM CoQ10 upregulates the sulfide oxidation pathway, restoring its function as well as other downstream and tangential effects of impaired sulfur metabolism [197].
Investigation of the mouse Coq7 homolog, Mclk1, showed that homozygous knockout resulted in embryonic lethality [198]. Heterozygous Mclk1+/− mice, however, live a normal lifespan with no significant defects in subcellular CoQ9 levels or mitochondrial morphology. Subcellular distribution of CoQ9 is altered, which may explain the lowered oxygen consumption and ATP production phenotype of heterozygous Mclk1+/− mitochondria [199,200]. Supplementation with CoQ10 significantly increases mitochondrial CoQ10 content and restores the electron transport chain defect [198].
Rare deleterious mutations in the human COQ9 gene result in severe multisystem disorders, affecting the central nervous system, cardiac, and renal systems [201]. Homozygous Coq9 mice (Coq9R239X/R239X) were prepared to model the human COQ9R244X deficiency [180]. These mice exhibit profound brain damage due to oxidative damage, neuronal apoptosis, and demyelination in the pons and medulla oblongata [180]. After two months of 240 mg/kg/day CoQ10 or CoQ10H2 oral supplementation, significant increases were detected in plasma, liver, and muscle tissue, followed by smaller increases in the heart, kidney, cerebrum, and cerebellum [180]. Supplementation with CoQ10H2 appeared to alleviate histopathological symptoms of CoQ deficiency in the brain, whereas CoQ10 did so to a lesser extent.
5. Conclusions
CoQ is an essential and ubiquitous lipid with many complex roles. However, there remains much to be learned about the mechanisms responsible for its uptake, intracellular trafficking, and distribution. Here, we have summarized the work in several model organisms used to investigate how CoQ is trafficked.
Yeast is an extraordinarily powerful model organism that has been used to identify proteins involved in the uptake and trafficking of exogenous CoQ6 to mitochondrial respiratory complexes. The yeast coq mutants completely lack CoQ6 biosynthesis and are thus respiratory deficient and fail to grow in a medium containing a non-fermentable carbon source. Supplementation with exogenous CoQ restores respiration and growth of coq mutants, and supplementation with the soluble CoQ2 analog can bypass defects in endocytosis and membrane trafficking [18,19,58]. Fernandez-del-Rio et al. [18] identified six genes that are essential for the uptake and trafficking of exogenous CoQ6 to mitochondrial respiratory complexes—RTS1, CDC10, RVS161, RVS167, VPS1, and NAT3. More work is needed to determine the role that each of these genes plays in the uptake and trafficking of exogenous CoQ6. While supplementation of ORFΔcoq2Δ double mutants with exogenous CoQ6 is a powerful approach for identifying genes required for CoQ6 uptake and trafficking, genes that are redundant or essential for respiration cannot be identified with exogenous CoQ6 rescue screens.
Yeast is also a powerful model for determining pathways and proteins responsible for the trafficking of endogenously synthesized CoQ6. Biosynthesis and transport of endogenously synthesized CoQ6 from the mitochondria in yeast is possibly mediated by ERMES [88,89]. However, direct transport of CoQ6 through ERMES has not yet been observed. Cqd1 and Cqd2 are mitochondrial inner membrane proteins that face the intermembrane space and influence the cellular distribution of endogenous CoQ6, and likely play a role in transporting CoQ6 from the mitochondrial inner membrane to the outer membrane and ER [93]. It is likely that other still unidentified CoQ6 transporters may contribute to trafficking.
Mammalian cell lines are an ideal model for investigating the cellular uptake and intracellular distribution of CoQ. Synthetic vehicles, such as cyclodextrin, caspofungin, micelles, liposomes, and nanodisks have been shown to dramatically improve the bioavailability of exogenous CoQ [117]. Moreover, supplementation with CoQ10H2 displays improved bioavailability in human dermal fibroblasts compared to CoQ10 [135], a trend that is also observed in rodent models [180,181]. Treatment of HL-60 cells with brefeldin-A shows that intracellular distribution of both endogenous and exogenous CoQ relies at least in part on the endomembrane system [83], though more work is needed to identify essential players of this pathway for CoQ transport. Recent work in HeLa cells has identified STARD7 as a phosphatidyl transfer protein involved in the export of endogenous CoQ from the mitochondria to the plasma membrane, where it plays a role in ferroptosis suppression [63]. Work in mouse endothelial brain cells has identified scavenger receptor SR-B1, receptor for advanced glycation endproducts (RAGE), and LRP-1 to be involved in transport of lipoprotein-associated CoQ10 across the blood–brain barrier [141]. CoQ10 may also be delivered to BAT cells by receptor-mediated uptake, via-the CD36 scavenger receptor [2,148].
Rodent models are useful for investigating the bioavailability of CoQ supplements and CoQ distribution in tissues of both wild-type animals as well as those harboring gene mutations that cause primary CoQ10 deficiency [6,160]. Dietary CoQ in rodents is taken up at the GI tract [164], and is then likely packaged into chylomicrons by intestinal enterocytes and transported into the lymphatic system, though more work needs to be performed to identify the pathways of endothelial uptake and to detect the supplemented CoQ in chylomicrons. CoQ from chylomicron remnants is likely repackaged with de novo CoQ at the liver and secreted in lipoprotein particles, with the majority in VLDL. Tissue localization of supplemented CoQ in rodents varies across studies and CoQ formulations, but enrichment is usually observed in the liver, plasma, and serum, with varying results in other tissues such as the heart, brain, and kidney. Supplemented CoQ is enriched in subcellular mitochondrial and light mitochondrial/lysosomal fractions. Moreover, transgenic mouse models have been used to investigate human mutations and canonical single nucleotide variants in Coq genes in order to evaluate the efficacy of treatment of CoQ-deficient mouse models with CoQ10 supplements.
It is important to note that comparisons across studies are challenging due to variability in CoQ formulations and dosage. Comparative studies of CoQ10 and CoQ10H2 must use proper precautions and controls to prevent rapid oxidation of CoQ10H2. Unfortunately, rodent studies that investigate CoQ10 uptake sometimes overlook endogenous CoQ9 content, despite CoQ9 being the predominant isoform in mice and rats. Many aspects of CoQ uptake and transport remain unknown or poorly understood. What is the relative importance of the endomembrane system and membrane contact sites in the transport of CoQ? How are CoQ transport and intracellular distribution of CoQ regulated? Elucidating the pathways of how organisms assimilate CoQ supplements, its cellular uptake, and pathways of intracellular distribution is important to understanding the functional roles of CoQ and may lead to improved therapeutics for exogenous supplementation.
Author Contributions
Writing—original draft preparation, A.J., K.A.A. and M.D.G.; writing—review and editing, A.J., C.F.C., K.A.A. and M.D.G.; visualization, A.J., C.F.C., K.A.A. and M.D.G.; supervision, C.F.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Greg Payne, Lucia Fernandez-del-Rio, Noelle Alexa Novales, Sining (Cindy) Wang, Kelsey Feustel, and other members of the C.F. Clarke lab for helpful advice and feedback on drafts of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
BAECs, bovine aortic endothelial cells; BAT, brown adipose tissue; BBB, blood–brain barrier; CoQ, coenzyme Q or ubiquinone; CoQH, ubisemiquinone; CoQH2, ubiquinol; CoQn, coenzyme Qn where n describes the number of isoprene units in the polyisoprenyl tail; COQ, denotes yeast or human gene required for biosynthesis of CoQ; Coq, denotes rodent genes; coq, denotes yeast gene harboring a mutation; COQ, denotes human or rodent polypeptide; Coq, denotes yeast polypeptide; CHDH, choline dehydrogenase; cyb5Red, NADH-cytochrome b5 reductase; DDMQH2, 2-methoxy-6-polyprenyl-1,4-benzohydroquinone; DHOD, dihydroorate dehydrogenase; DHP, 4,5-dihydroxy-3-polyprenylphenol; DHPB, 4,5-dihydroxy-3-polyprenylbenzoic acid; DMAPP, dimethylallyl pyrophosphate; DMQH2, 2-methoxy-5-methyl-6-polyprenyl-1,4-benzohydroquinone; DMeQH2, 3-methyl-6-methoxy-2-polyprenyl-1,4,5-benzenetriol; DMGDH, dimethylglycine dehydrogenase; ERMES, endoplasmic reticulum encounter structure; ETF, electron transfer flavoprotein; ETFDH, electron transfer flavoprotein dehydrogenase; FSP1, ferroptosis suppressor protein 1; GPDH, glycerol-3-phosphate dehydrogenase; GPX4, glutathione peroxidase 4; HAB, 3-hexaprenyl-4-aminobenzoic acid; 4-HB, 4-hydroxybenzoic acid; 4-Hbz, 4-hydroxybenzaldehyde; HDL, high-density lipoprotein; 4-HMA, 4-hydroxymandelate; HMPB, 4-hydroxy-5-methoxy-3-polyprenylbenzoic acid; HPB, 4-hydroxy-3-polyprenyl-benzoic acid; 4-HPP, 4-hydroxyphenylpyruvate; HUVECs, human umbilical vein endothelial cells; IPP, isopentenyl pyrophosphate; LDL, low-density lipoprotein; LRP-1, LDL receptor related protein 1; MEFs, mouse embryonic fibroblasts; MICOS, mitochondrial contact site and cristae organization system; NPC1L1, Niemann–Pick C1-like 1; NQO1, NAD(P)H:quinone oxidoreductase 1; pABA, para-aminobenzoic acid; PBEC, porcine brain endothelial cells; PMRS, plasma membrane redox system; PRDH, proline dehydrogenase; PUFAs, polyunsaturated fatty acids; RAGE, receptor for advanced glycation end products; SARDH, sarcosine dehydrogenase; SQR, sulfide:quinone oxidoreductase; SR-B1, scavenger receptor; VLDL, very low-density lipoprotein.
References
- Arenas-Jal, M.; Sune-Negre, J.M.; Garcia-Montoya, E. Coenzyme Q10 supplementation: Efficacy, safety, and formulation challenges. Compr. Rev. Food Sci. Food Saf. 2020, 19, 574–594. [Google Scholar] [CrossRef][Green Version]
- Mantle, D.; Dybring, A. Bioavailability of Coenzyme Q(10): An Overview of the Absorption Process and Subsequent Metabolism. Antioxidants 2020, 9, 386. [Google Scholar] [CrossRef]
- Pravst, I.; Rodriguez Aguilera, J.C.; Cortes Rodriguez, A.B.; Jazbar, J.; Locatelli, I.; Hristov, H.; Zmitek, K. Comparative Bioavailability of Different Coenzyme Q10 Formulations in Healthy Elderly Individuals. Nutrients 2020, 12, 784. [Google Scholar] [CrossRef][Green Version]
- Mantle, D.; Turton, N.; Hargreaves, I.P. Depletion and Supplementation of Coenzyme Q10 in Secondary Deficiency Disorders. Front. Biosci. Landmark Ed. 2022, 27, 322. [Google Scholar] [CrossRef] [PubMed]
- Sifuentes-Franco, S.; Sanchez-Macias, D.C.; Carrillo-Ibarra, S.; Rivera-Valdes, J.J.; Zuniga, L.Y.; Sanchez-Lopez, V.A. Antioxidant and Anti-Inflammatory Effects of Coenzyme Q10 Supplementation on Infectious Diseases. Healthcare 2022, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. The efficacy of coenzyme Q(10) treatment in alleviating the symptoms of primary coenzyme Q(10) deficiency: A systematic review. J. Cell Mol. Med. 2022, 26, 4635–4644. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lluch, G.; Del Pozo-Cruz, J.; Sanchez-Cuesta, A.; Cortes-Rodriguez, A.B.; Navas, P. Bioavailability of coenzyme Q10 supplements depends on carrier lipids and solubilization. Nutrition 2019, 57, 133–140. [Google Scholar] [CrossRef]
- Miles, M.V. The uptake and distribution of coenzyme Q10. Mitochondrion 2007, 7, S72–S77. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Lopez-Lluch, G.; Hargreaves, I.P. Coenzyme Q10 Metabolism: A Review of Unresolved Issues. Int. J. Mol. Sci. 2023, 24, 2585. [Google Scholar] [CrossRef]
- Kawamukai, M. Biosynthesis of coenzyme Q in eukaryotes. Biosci. Biotechnol. Biochem. 2016, 80, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Pierrel, F.; Burgardt, A.; Lee, J.H.; Pelosi, L.; Wendisch, V.F. Recent advances in the metabolic pathways and microbial production of coenzyme Q. World J. Microbiol. Biotechnol. 2022, 38, 58. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Molecular genetics of ubiquinone biosynthesis in animals. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 69–88. [Google Scholar] [CrossRef][Green Version]
- Hayashi, K.; Ogiyama, Y.; Yokomi, K.; Nakagawa, T.; Kaino, T.; Kawamukai, M. Functional Conservation of Coenzyme Q Biosynthetic Genes among Yeasts, Plants, and Humans. PLoS ONE 2014, 9, e99038. [Google Scholar] [CrossRef][Green Version]
- Aussel, L.; Pierrel, F.; Loiseau, L.; Lombard, M.; Fontecave, M.; Barras, F. Biosynthesis and physiology of coenzyme Q in bacteria. Biochim. Biophys. Acta 2014, 1837, 1004–1011. [Google Scholar] [CrossRef]
- Ramasarma, T. Natural Occurrence and Distribution of Coenzyme Q. In Coenzyme Q Biochemistry, Bioenergetics and Clinical Applications of Ubiquinone; Lenaz, G., Ed.; John Wiley & Sons: New York, NY, USA, 1985; pp. 67–81. [Google Scholar]
- Quinn, P.J. Lipid-lipid interactions in bilayer membranes: Married couples and casual liaisons. Prog. Lipid Res. 2012, 51, 179–198. [Google Scholar] [CrossRef]
- Braasch-Turi, M.M.; Koehn, J.T.; Crans, D.C. Chemistry of Lipoquinones: Properties, Synthesis, and Membrane Location of Ubiquinones, Plastoquinones, and Menaquinones. Int. J. Mol. Sci. 2022, 23, 12856. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Del-Rio, L.; Kelly, M.E.; Contreras, J.; Bradley, M.C.; James, A.M.; Murphy, M.P.; Payne, G.S.; Clarke, C.F. Genes and lipids that impact uptake and assimilation of exogenous coenzyme Q in Saccharomyces cerevisiaeae. Free Radic. Biol. Med. 2020, 154, 105–118. [Google Scholar] [CrossRef] [PubMed]
- James, A.M.; Cocheme, H.M.; Murai, M.; Miyoshi, H.; Murphy, M.P. Complementation of coenzyme Q-deficient yeast by coenzyme Q analogues requires the isoprenoid side chain. FEBS J. 2010, 277, 2067–2082. [Google Scholar] [CrossRef]
- Alcazar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef]
- Pallotti, F.; Bergamini, C.; Lamperti, C.; Fato, R. The Roles of Coenzyme Q in Disease: Direct and Indirect Involvement in Cellular Functions. Int. J. Mol. Sci. 2021, 23, 128. [Google Scholar] [CrossRef]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2022, 23, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Purhonen, J.; Kallijarvi, J. The mitochondrial coenzyme Q junction and complex III: Biochemistry and pathophysiology. FEBS J. 2021, 289, 6936–6958. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L.; Forsmark-Andree, P. Ubiquinol: An endogenous antioxidant in aerobic organisms. Clin. Investig. 1993, 71, S60–S65. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Yubero-Serrano, E.M.; Villalba, J.M.; Lopez-Miranda, J. Coenzyme Q(10): From bench to clinic in aging diseases, a translational review. Crit. Rev. Food Sci. Nutr. 2019, 59, 2240–2257. [Google Scholar] [CrossRef]
- Kagan, V.E.; Fabisiak, J.P.; Quinn, P.J. Coenzyme Q and vitamin E need each other as antioxidants. Protoplasma 2000, 214, 11–18. [Google Scholar] [CrossRef]
- Do, Q.; Zhang, R.; Hooper, G.; Xu, L. Differential Contributions of Distinct Free Radical Peroxidation Mechanisms to the Induction of Ferroptosis. J. Am. Chem. Soc. 2023, 3, 1100–1117. [Google Scholar] [CrossRef] [PubMed]
- Hyun, D.H.; Hernandez, J.O.; Mattson, M.P.; de Cabo, R. The plasma membrane redox system in aging. Ageing Res. Rev. 2006, 5, 209–220. [Google Scholar] [CrossRef]
- Baschiera, E.; Sorrentino, U.; Calderan, C.; Desbats, M.A.; Salviati, L. The multiple roles of coenzyme Q in cellular homeostasis and their relevance for the pathogenesis of coenzyme Q deficiency. Free. Radic. Biol. Med. 2021, 166, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Villalba, J.M.; Navas, P. Plasma membrane redox system in the control of stress-induced apoptosis. Antioxid. Redox Signal. 2000, 2, 213–230. [Google Scholar] [CrossRef]
- Martin, S.F.; Gomez-Diaz, C.; Bello, R.I.; Navas, P.; Villalba, J.M. Inhibition of neutral Mg2+-dependent sphingomyelinase by ubiquinol-mediated plasma membrane electron transport. Protoplasma 2003, 221, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Ito, J.; Wu, Z.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 2022, 608, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef][Green Version]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guerra, R.M.; Pagliarini, D.J. Coenzyme Q biochemistry and biosynthesis. Trends Biochem. Sci. 2023, 48, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.M.; Bradley, M.C.; Fernandez-Del-Rio, L.; Nag, A.; Tsui, H.S.; Clarke, C.F. Coenzyme Q10 deficiencies: Pathways in yeast and humans. Essays Biochem. 2018, 62, 361–376. [Google Scholar] [CrossRef][Green Version]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Robinson, K.P.; Jochem, A.; Johnson, S.E.; Reddy, T.R.; Russell, J.D.; Coon, J.J.; Pagliarini, D.J. Defining intermediates and redundancies in coenzyme Q precursor biosynthesis. J. Biol. Chem. 2021, 296, 100643. [Google Scholar] [CrossRef]
- Banh, R.S.; Kim, E.S.; Spillier, Q.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Shi, G.; Jones, D.R.; Kimmelman, A.C.; Pacold, M.E. The polar oxy-metabolome reveals the 4-hydroxymandelate CoQ10 synthesis pathway. Nature 2021, 597, 420–425. [Google Scholar] [CrossRef]
- Valera, M.J.; Boido, E.; Ramos, J.C.; Manta, E.; Radi, R.; Dellacassa, E.; Carrau, F. The Mandelate Pathway, an Alternative to the Phenylalanine Ammonia Lyase Pathway for the Synthesis of Benzenoids in Ascomycete Yeasts. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- Salviati, L.; Trevisson, E.; Doimo, M.; Navas, P. Primary Coenzyme Q(10) Deficiency. In GeneReviews((R)); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Wang, S.; Jain, A.; Novales, N.A.; Nashner, A.N.; Tran, F.; Clarke, C.F. Predicting and Understanding the Pathology of Single Nucleotide Variants in Human COQ Genes. Antioxidants 2022, 11, 2308. [Google Scholar] [CrossRef]
- Quinzii, C.M.; Hirano, M. Primary and secondary CoQ(10) deficiencies in humans. BioFactors 2011, 37, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008, 358, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Pierrel, F. Impact of Chemical Analogs of 4-Hydroxybenzoic Acid on Coenzyme Q Biosynthesis: From Inhibition to Bypass of Coenzyme Q Deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Herebian, D.; Lopez, L.C.; Distelmaier, F. Bypassing human CoQ(10) deficiency. Mol. Genet. Metab. 2018, 123, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Smith, C.; Parboosingh, J.S.; Khan, A.; Innes, M.; Hekimi, S. Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J. Cell Mol. Med. 2017, 21, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Luna-Sanchez, M.; Diaz-Casado, E.; Barca, E.; Tejada, M.A.; Montilla-Garcia, A.; Cobos, E.J.; Escames, G.; Acuna-Castroviejo, D.; Quinzii, C.M.; Lopez, L.C. The clinical heterogeneity of coenzyme Q10 deficiency results from genotypic differences in the Coq9 gene. EMBO Mol. Med. 2015, 7, 670–687. [Google Scholar] [CrossRef] [PubMed]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocana, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta-Mol. Basis Dis. 2014, 1842, 1–6. [Google Scholar] [CrossRef][Green Version]
- Gomez, F.; Saiki, R.; Chin, R.; Srinivasan, C.; Clarke, C.F. Restoring de novo coenzyme Q biosynthesis in Caenorhabditis elegans coq-3 mutants yields profound rescue compared to exogenous coenzyme Q supplementation. Gene 2012, 506, 106–116. [Google Scholar] [CrossRef][Green Version]
- Duina, A.A.; Miller, M.E.; Keeney, J.B. Budding yeast for budding geneticists: A primer on the Saccharomyces cerevisiaeae model system. Genetics 2014, 197, 33–48. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nielsen, J. Yeast Systems Biology: Model Organism and Cell Factory. Biotechnol. J. 2019, 14, e1800421. [Google Scholar] [CrossRef][Green Version]
- Jonassen, T.; Proft, M.; Randez-Gil, F.; Schultz, J.R.; Marbois, B.N.; Entian, K.D.; Clarke, C.F. Yeast Clk-1 homologue (Coq7/Cat5) is a mitochondrial protein in coenzyme Q synthesis. J. Biol. Chem. 1998, 273, 3351–3357. [Google Scholar] [CrossRef][Green Version]
- Do, T.Q.; Hsu, A.Y.; Jonassen, T.; Lee, P.T.; Clarke, C.F. A defect in coenzyme Q biosynthesis is responsible for the respiratory deficiency in Saccharomyces cerevisiaeae abc1 mutants. J. Biol. Chem. 2001, 276, 18161–18168. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Padilla-López, S.; Jiménez-Hidalgo, M.; Martín-Montalvo, A.; Clarke, C.F.; Navas, P.; Santos-Ocaña, C. Genetic evidence for the requirement of the endocytic pathway in the uptake of coenzyme Q6 in Saccharomyces cerevisiaeae. Biochim. Biophys. Acta BBA—Biomembr. 2009, 1788, 1238–1248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- James, A.M.; Cocheme, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef][Green Version]
- Barros, M.H.; Johnson, A.; Gin, P.; Marbois, B.N.; Clarke, C.F.; Tzagoloff, A. The Saccharomyces cerevisiae COQ10 gene encodes a START domain protein required for function of coenzyme Q in respiration. J. Biol. Chem. 2005, 280, 42627–42635. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Allan, C.M.; Hill, S.; Morvaridi, S.; Saiki, R.; Johnson, J.S.; Liau, W.S.; Hirano, K.; Kawashima, T.; Ji, Z.; Loo, J.A.; et al. A conserved START domain coenzyme Q-binding polypeptide is required for efficient Q biosynthesis, respiratory electron transport, and antioxidant function in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2013, 1831, 776–791. [Google Scholar] [CrossRef][Green Version]
- Busso, C.; Tahara, E.B.; Ogusucu, R.; Augusto, O.; Ferreira-Junior, J.R.; Tzagoloff, A.; Kowaltowski, A.J.; Barros, M.H. Saccharomyces cerevisiaeae coq10 null mutants are responsive to antimycin A. FEBS J. 2010, 277, 4530–4538. [Google Scholar] [CrossRef][Green Version]
- Tsui, H.S.; Pham, N.V.B.; Amer, B.R.; Bradley, M.C.; Gosschalk, J.E.; Gallagher-Jones, M.; Ibarra, H.; Clubb, R.T.; Blaby-Haas, C.E.; Clarke, C.F. Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function. J. Lipid Res. 2019, 60, 1293–1310. [Google Scholar] [CrossRef]
- Deshwal, S.; Onishi, M.; Tatsuta, T.; Bartsch, T.; Cors, E.; Ried, K.; Lemke, K.; Nolte, H.; Giavalisco, P.; Langer, T. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat. Cell Biol. 2023, 25, 246–257. [Google Scholar] [CrossRef]
- Jin, G.; Horinouchi, R.; Sagawa, T.; Orimo, N.; Kubo, H.; Yoshimura, S.; Fujisawa, A.; Kashiba, M.; Yamamoto, Y. Coenzyme Q10-Binding/Transfer Protein Saposin B also Binds gamma-Tocopherol. J. Clin. Biochem. Nutr. 2008, 43, 95–100. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jin, G.; Kubo, H.; Kashiba, M.; Horinouchi, R.; Hasegawa, M.; Suzuki, M.; Sagawa, T.; Oizumi, M.; Fujisawa, A.; Tsukamoto, H.; et al. Saposin B is a human coenzyme q10-binding/transfer protein. J. Clin. Biochem. Nutr. 2008, 42, 167–174. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arino, J.; Velazquez, D.; Casamayor, A. Ser/Thr protein phosphatases in fungi: Structure, regulation and function. Microb. Cell 2019, 6, 217–256. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Han, Q.; Pan, C.; Wang, Y.; Wang, N.; Wang, Y.; Sang, J. The PP2A regulatory subunits, Cdc55 and Rts1, play distinct roles in Candida albicans’ growth, morphogenesis, and virulence. Fungal Genet. Biol. 2019, 131, 103240. [Google Scholar] [CrossRef]
- Glomb, O.; Gronemeyer, T. Septin Organization and Functions in Budding Yeast. Front. Cell Dev. Biol. 2016, 4, 123. [Google Scholar] [CrossRef][Green Version]
- Juanes, M.A.; Piatti, S. The final cut: Cell polarity meets cytokinesis at the bud neck in S. cerevisiae. Cell Mol. Life Sci. CMLS 2016, 73, 3115–3136. [Google Scholar] [CrossRef][Green Version]
- Renz, C.; Oeljeklaus, S.; Grinhagens, S.; Warscheid, B.; Johnsson, N.; Gronemeyer, T. Identification of Cell Cycle Dependent Interaction Partners of the Septins by Quantitative Mass Spectrometry. PLoS ONE 2016, 11, e0148340. [Google Scholar] [CrossRef]
- Song, K.; Russo, G.; Krauss, M. Septins As Modulators of Endo-Lysosomal Membrane Traffic. Front. Cell Dev. Biol. 2016, 4, 124. [Google Scholar] [CrossRef][Green Version]
- Youn, J.Y.; Friesen, H.; Kishimoto, T.; Henne, W.M.; Kurat, C.F.; Ye, W.; Ceccarelli, D.F.; Sicheri, F.; Kohlwein, S.D.; McMahon, H.T.; et al. Dissecting BAR domain function in the yeast Amphiphysins Rvs161 and Rvs167 during endocytosis. Mol. Biol. Cell 2010, 21, 3054–3069. [Google Scholar] [CrossRef][Green Version]
- Smaczynska-de, R., II; Allwood, E.G.; Mishra, R.; Booth, W.I.; Aghamohammadzadeh, S.; Goldberg, M.W.; Ayscough, K.R. Yeast dynamin Vps1 and amphiphysin Rvs167 function together during endocytosis. Traffic 2012, 13, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Kim, K. From membranes to organelles: Emerging roles for dynamin-like proteins in diverse cellular processes. Eur. J. Cell Biol. 2014, 93, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef] [PubMed]
- Aksnes, H.; Hole, K.; Arnesen, T. Molecular, cellular, and physiological significance of N-terminal acetylation. Int. Rev. Cell Mol. Biol. 2015, 316, 267–305. [Google Scholar] [CrossRef]
- Feyder, S.; De Craene, J.O.; Bar, S.; Bertazzi, D.L.; Friant, S. Membrane trafficking in the yeast Saccharomyces cerevisiaeae model. Int. J. Mol. Sci. 2015, 16, 1509–1525. [Google Scholar] [CrossRef][Green Version]
- Wei, Z.; Su, W.; Lou, H.; Duan, S.; Chen, G. Trafficking pathway between plasma membrane and mitochondria via clathrin-mediated endocytosis. J. Mol. Cell Biol. 2018, 10, 539–548. [Google Scholar] [CrossRef][Green Version]
- Ellenrieder, L.; Rampelt, H.; Becker, T. Connection of Protein Transport and Organelle Contact Sites in Mitochondria. J. Mol. Biol. 2017, 429, 2148–2160. [Google Scholar] [CrossRef]
- Zung, N.; Schuldiner, M. New horizons in mitochondrial contact site research. Biol. Chem. 2020, 401, 793–809. [Google Scholar] [CrossRef]
- Elbaz-Alon, Y.; Rosenfeld-Gur, E.; Shinder, V.; Futerman, A.H.; Geiger, T.; Schuldiner, M. A dynamic interface between vacuoles and mitochondria in yeast. Dev. Cell 2014, 30, 95–102. [Google Scholar] [CrossRef][Green Version]
- Santos-Ocana, C.; Do, T.Q.; Padilla, S.; Navas, P.; Clarke, C.F. Uptake of exogenous coenzyme Q and transport to mitochondria is required for bc1 complex stability in yeast coq mutants. J. Biol. Chem. 2002, 277, 10973–10981. [Google Scholar] [CrossRef][Green Version]
- Fernandez-Ayala, D.J.; Brea-Calvo, G.; Lopez-Lluch, G.; Navas, P. Coenzyme Q distribution in HL-60 human cells depends on the endomembrane system. Biochim. Biophys. Acta 2005, 1713, 129–137. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wozny, M.R.; Di Luca, A.; Morado, D.R.; Picco, A.; Khaddaj, R.; Campomanes, P.; Ivanovic, L.; Hoffmann, P.C.; Miller, E.A.; Vanni, S.; et al. In situ architecture of the ER-mitochondria encounter structure. Nature 2023, 618, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Tirrell, P.S.; Nguyen, K.N.; Luby-Phelps, K.; Friedman, J.R. MICOS subcomplexes assemble independently on the mitochondrial inner membrane in proximity to ER contact sites. J. Cell Biol. 2020, 219, e202003024. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Tamura, Y.; Kojima, R.; Bala, S.; Asai, E.; Michel, A.H.; Kornmann, B.; Riezman, I.; Riezman, H.; Sakae, Y.; et al. Structure-function insights into direct lipid transfer between membranes by Mmm1-Mdm12 of ERMES. J. Cell Biol. 2018, 217, 959–974. [Google Scholar] [CrossRef]
- Subramanian, K.; Jochem, A.; Le Vasseur, M.; Lewis, S.; Paulson, B.R.; Reddy, T.R.; Russell, J.D.; Coon, J.J.; Pagliarini, D.J.; Nunnari, J. Coenzyme Q biosynthetic proteins assemble in a substrate-dependent manner into domains at ER-mitochondria contacts. J. Cell Biol. 2019, 218, 1353–1369. [Google Scholar] [CrossRef][Green Version]
- Eisenberg-Bord, M.; Tsui, H.S.; Antunes, D.; Fernandez-Del-Rio, L.; Bradley, M.C.; Dunn, C.D.; Nguyen, T.P.T.; Rapaport, D.; Clarke, C.F.; Schuldiner, M. The Endoplasmic Reticulum-Mitochondria Encounter Structure Complex Coordinates Coenzyme Q Biosynthesis. Contact 2019, 2, 2515256418825409. [Google Scholar] [CrossRef][Green Version]
- Leonzino, M.; Reinisch, K.M.; De Camilli, P. Insights into VPS13 properties and function reveal a new mechanism of eukaryotic lipid transport. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2021, 1866, 159003. [Google Scholar] [CrossRef]
- John Peter, A.T.; Herrmann, B.; Antunes, D.; Rapaport, D.; Dimmer, K.S.; Kornmann, B. Vps13-Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J. Cell Biol. 2017, 216, 3219–3229. [Google Scholar] [CrossRef][Green Version]
- Adlakha, J.; Hong, Z.; Li, P.; Reinisch, K.M. Structural and biochemical insights into lipid transport by VPS13 proteins. J. Cell Biol. 2022, 221, e202202030. [Google Scholar] [CrossRef]
- Kemmerer, Z.A.; Robinson, K.P.; Schmitz, J.M.; Manicki, M.; Paulson, B.R.; Jochem, A.; Hutchins, P.D.; Coon, J.J.; Pagliarini, D.J. UbiB proteins regulate cellular CoQ distribution in Saccharomyces cerevisiaeae. Nat. Commun. 2021, 12, 4769. [Google Scholar] [CrossRef] [PubMed]
- Reidenbach, A.G.; Kemmerer, Z.A.; Aydin, D.; Jochem, A.; McDevitt, M.T.; Hutchins, P.D.; Stark, J.L.; Stefely, J.A.; Reddy, T.; Hebert, A.S.; et al. Conserved Lipid and Small-Molecule Modulation of COQ8 Reveals Regulation of the Ancient Kinase-like UbiB Family. Cell Chem. Biol. 2018, 25, 154–165.e111. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vazquez-Fonseca, L.; Schaefer, J.; Navas-Enamorado, I.; Santos-Ocana, C.; Hernandez-Camacho, J.D.; Guerra, I.; Cascajo, M.V.; Sanchez-Cuesta, A.; Horvath, Z.; Siendones, E.; et al. ADCK2 Haploinsufficiency Reduces Mitochondrial Lipid Oxidation and Causes Myopathy Associated with CoQ Deficiency. J. Clin. Med. 2019, 8, 1374. [Google Scholar] [CrossRef][Green Version]
- Khosravi, S.; Chelius, X.; Unger, A.K.; Rieger, D.; Frickel, J.; Sachsenheimer, T.; Luchtenborg, C.; Schieweck, R.; Brugger, B.; Westermann, B.; et al. The UbiB family member Cqd1 forms a novel membrane contact site in mitochondria. J. Cell Sci. 2023, 136, jcs260578. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ocana, C.; Cordoba, F.; Crane, F.L.; Clarke, C.F.; Navas, P. Coenzyme Q6 and iron reduction are responsible for the extracellular ascorbate stabilization at the plasma membrane of Saccharomyces cerevisiaeae. J. Biol. Chem. 1998, 273, 8099–8105. [Google Scholar] [CrossRef][Green Version]
- Gomez-Diaz, C.; Villalba, J.M.; Perez-Vicente, R.; Crane, F.L.; Navas, P. Ascorbate stabilization is stimulated in rho(0)HL-60 cells by CoQ10 increase at the plasma membrane. Biochem. Biophys. Res. Commun. 1997, 234, 79–81. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. The diverse functionality of NQO1 and its roles in redox control. Redox Biol. 2021, 41, 101950. [Google Scholar] [CrossRef]
- Gomez-Diaz, C.; Rodriguez-Aguilera, J.C.; Barroso, M.P.; Villalba, J.M.; Navarro, F.; Crane, F.L.; Navas, P. Antioxidant ascorbate is stabilized by NADH-coenzyme Q10 reductase in the plasma membrane. J. Bioenerg. Biomembr. 1997, 29, 251–257. [Google Scholar] [CrossRef]
- Santos-Ocana, C.; Cascajo, M.V.; Alcazar-Fabra, M.; Staiano, C.; Lopez-Lluch, G.; Brea-Calvo, G.; Navas, P. Cellular Models for Primary CoQ Deficiency Pathogenesis Study. Int. J. Mol. Sci. 2021, 22, 10211. [Google Scholar] [CrossRef]
- Frontinan-Rubio, J.; Llanos-Gonzalez, E.; Garcia-Carpintero, S.; Peinado, J.R.; Ballesteros-Yanez, I.; Rayo, M.V.; de la Fuente, J.; Perez-Garcia, V.M.; Perez-Romasanta, L.A.; Malumbres, M.; et al. CoQ(10) reduces glioblastoma growth and infiltration through proteome remodeling and inhibition of angiogenesis and inflammation. Cell Oncol. 2023, 46, 65–77. [Google Scholar] [CrossRef]
- Petrillo, S.; D’Amico, J.; Nicita, F.; Torda, C.; Vasco, G.; Bertini, E.S.; Cappa, M.; Piemonte, F. Antioxidant Response in Human X-Linked Adrenoleukodystrophy Fibroblasts. Antioxidants 2022, 11, 2125. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Lei, G.; Zhang, Y.; Yan, Y.; Mao, C.; Kondiparthi, L.; Shi, J.; Liu, X.; Horbath, A.; Das, M.; et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 2022, 13, 2206. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Tsui, M.G.; Tsang, J.K.W.; Goit, R.K.; Yao, K.M.; So, K.F.; Lam, W.C.; Lo, A.C.Y. Involvement of FSP1-CoQ(10)-NADH and GSH-GPx-4 pathways in retinal pigment epithelium ferroptosis. Cell Death Dis. 2022, 13, 468. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.A.; Kindermann, B.; Althammer, M.; Klapper, M.; Vormann, J.; Littarru, G.P.; Doring, F. Coenzyme Q10 affects expression of genes involved in cell signalling, metabolism and transport in human CaCo-2 cells. Int. J. Biochem. Cell Biol. 2005, 37, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Liu, M.; Portincasa, P.; Wang, D.Q. New insights into the molecular mechanism of intestinal fatty acid absorption. Eur. J. Clin. Investig. 2013, 43, 1203–1223. [Google Scholar] [CrossRef][Green Version]
- Kiyose, C. Absorption, transportation, and distribution of vitamin E homologs. Free Radic. Biol. Med. 2021, 177, 226–237. [Google Scholar] [CrossRef]
- Nakano, T.; Inoue, I.; Murakoshi, T. A Newly Integrated Model for Intestinal Cholesterol Absorption and Efflux Reappraises How Plant Sterol Intake Reduces Circulating Cholesterol Levels. Nutrients 2019, 11, 310. [Google Scholar] [CrossRef][Green Version]
- Degim, Z.; Unal, N.; Essiz, D.; Abbasoglu, U. The effect of various liposome formulations on insulin penetration across Caco-2 cell monolayer. Life Sci. 2004, 75, 2819–2827. [Google Scholar] [CrossRef]
- Kamiloglu, S.; Capanoglu, E.; Grootaert, C.; Van Camp, J. Anthocyanin Absorption and Metabolism by Human Intestinal Caco-2 Cells—A Review. Int. J. Mol. Sci. 2015, 16, 21555–21574. [Google Scholar] [CrossRef][Green Version]
- Song, K.H.; Chung, S.J.; Shim, C.K. Enhanced intestinal absorption of salmon calcitonin (sCT) from proliposomes containing bile salts. J. Control. Release 2005, 106, 298–308. [Google Scholar] [CrossRef]
- Lea, T. Caco-2 Cell Line. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., Lopez-Exposito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 103–111. [Google Scholar]
- Navas, P.; Fernandez-Ayala, D.M.; Martin, S.F.; Lopez-Lluch, G.; De Caboa, R.; Rodriguez-Aguilera, J.C.; Villalba, J.M. Ceramide-dependent caspase 3 activation is prevented by coenzyme Q from plasma membrane in serum-deprived cells. Free Radic. Res. 2002, 36, 369–374. [Google Scholar] [CrossRef]
- Bhagavan, H.N.; Chopra, R.K.; Craft, N.E.; Chitchumroonchokchai, C.; Failla, M.L. Assessment of coenzyme Q10 absorption using an in vitro digestion-Caco-2 cell model. Int. J. Pharm. 2007, 333, 112–117. [Google Scholar] [CrossRef]
- Saokham, P.; Loftsson, T. gamma-Cyclodextrin. Int. J. Pharm. 2017, 516, 278–292. [Google Scholar] [CrossRef]
- Pastor-Maldonado, C.J.; Suarez-Rivero, J.M.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Munuera-Cabeza, M.; Suarez-Carrillo, A.; Talaveron-Rey, M.; Sanchez-Alcazar, J.A. Coenzyme Q(10): Novel Formulations and Medical Trends. Int. J. Mol. Sci. 2020, 21, 8432. [Google Scholar] [CrossRef] [PubMed]
- Failla, M.L.; Chitchumroonchokchai, C.; Aoki, F. Increased bioavailability of ubiquinol compared to that of ubiquinone is due to more efficient micellarization during digestion and greater GSH-dependent uptake and basolateral secretion by Caco-2 cells. J. Agric. Food Chem. 2014, 62, 7174–7182. [Google Scholar] [CrossRef] [PubMed]
- Burger, N.; Logan, A.; Prime, T.A.; Mottahedin, A.; Caldwell, S.T.; Krieg, T.; Hartley, R.C.; James, A.M.; Murphy, M.P. A sensitive mass spectrometric assay for mitochondrial CoQ pool redox state in vivo. Free Radic. Biol. Med. 2020, 147, 37–47. [Google Scholar] [CrossRef]
- Naguib, Y.W.; Saha, S.; Skeie, J.M.; Acri, T.; Ebeid, K.; Abdel-Rahman, S.; Kesh, S.; Schmidt, G.A.; Nishimura, D.Y.; Banas, J.A.; et al. Solubilized ubiquinol for preserving corneal function. Biomaterials 2021, 275, 120842. [Google Scholar] [CrossRef]
- Xia, S.; Xu, S.; Zhang, X.; Zhong, F.; Wang, Z. Nanoliposomes mediate coenzyme Q10 transport and accumulation across human intestinal Caco-2 cell monolayer. J. Agric. Food Chem. 2009, 57, 7989–7996. [Google Scholar] [CrossRef]
- Hu, M.; Yang, F.; Huang, Y.; You, X.; Liu, D.; Sun, S.; Sui, S.F. Structural insights into the mechanism of human NPC1L1-mediated cholesterol uptake. Sci. Adv. 2021, 7, eabg3188. [Google Scholar] [CrossRef] [PubMed]
- Narushima, K.; Takada, T.; Yamanashi, Y.; Suzuki, H. Niemann-pick C1-like 1 mediates alpha-tocopherol transport. Mol. Pharmacol. 2008, 74, 42–49. [Google Scholar] [CrossRef]
- Nashimoto, S.; Takekawa, Y.; Takekuma, Y.; Sugawara, M.; Sato, Y. Transport via Niemann-Pick C1 Like 1 contributes to the intestinal absorption of ubiquinone. Drug Metab. Pharmacokinet. 2020, 35, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Moschetti, A.; Vine, L.N.; Lethcoe, K.; Dagda, R.K.; Ellison, P.; Ryan, R.O. Assembly and Characterization of Biocompatible Coenzyme Q(10) -Enriched Lipid Nanoparticles. Lipids 2020, 55, 141–149. [Google Scholar] [CrossRef]
- Moschetti, A.; Dagda, R.K.; Ryan, R.O. Coenzyme Q nanodisks counteract the effect of statins on C2C12 myotubes. Nanomedicine 2021, 37, 102439. [Google Scholar] [CrossRef]
- Fox, C.A.; Moschetti, A.; Ryan, R.O. Reconstituted HDL as a therapeutic delivery device. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 159025. [Google Scholar] [CrossRef]
- Suo, Z.; Fang, C.; Crawford, F.; Mullan, M. Superoxide free radical and intracellular calcium mediate A beta(1-42) induced endothelial toxicity. Brain Res. 1997, 762, 144–152. [Google Scholar] [CrossRef]
- Duran-Prado, M.; Frontinan, J.; Santiago-Mora, R.; Peinado, J.R.; Parrado-Fernandez, C.; Gomez-Almagro, M.V.; Moreno, M.; Lopez-Dominguez, J.A.; Villalba, J.M.; Alcain, F.J. Coenzyme Q10 protects human endothelial cells from beta-amyloid uptake and oxidative stress-induced injury. PLoS ONE 2014, 9, e109223. [Google Scholar] [CrossRef]
- Dhanasekaran, A.; Kotamraju, S.; Kalivendi, S.V.; Matsunaga, T.; Shang, T.; Keszler, A.; Joseph, J.; Kalyanaraman, B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J. Biol. Chem. 2004, 279, 37575–37587. [Google Scholar] [CrossRef][Green Version]
- Wani, M.R.; Shadab, G. Coenzyme Q10 protects isolated human blood cells from TiO(2) nanoparticles induced oxidative/antioxidative imbalance, hemolysis, cytotoxicity, DNA damage and mitochondrial impairment. Mol. Biol. Rep. 2021, 48, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.M.; Yang, T.Y.; Huang, H.C. Nicotinamide Mononucleotide and Coenzyme Q10 Protects Fibroblast Senescence Induced by Particulate Matter Preconditioned Mast Cells. Int. J. Mol. Sci. 2022, 23, 7539. [Google Scholar] [CrossRef]
- Gokce, E.H.; Korkmaz, E.; Tuncay-Tanriverdi, S.; Dellera, E.; Sandri, G.; Bonferoni, M.C.; Ozer, O. A comparative evaluation of coenzyme Q10-loaded liposomes and solid lipid nanoparticles as dermal antioxidant carriers. Int. J. Nanomed. 2012, 7, 5109–5117. [Google Scholar] [CrossRef][Green Version]
- Marcheggiani, F.; Kordes, S.; Cirilli, I.; Orlando, P.; Silvestri, S.; Vogelsang, A.; Moller, N.; Blatt, T.; Weise, J.M.; Damiani, E.; et al. Anti-ageing effects of ubiquinone and ubiquinol in a senescence model of human dermal fibroblasts. Free Radic. Biol. Med. 2021, 165, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Muta-Takada, K.; Terada, T.; Yamanishi, H.; Ashida, Y.; Inomata, S.; Nishiyama, T.; Amano, S. Coenzyme Q10 protects against oxidative stress-induced cell death and enhances the synthesis of basement membrane components in dermal and epidermal cells. BioFactors 2009, 35, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Staufer, T.; Schulze, M.L.; Schmutzler, O.; Kornig, C.; Welge, V.; Burkhardt, T.; Vietzke, J.P.; Vogelsang, A.; Weise, J.M.; Blatt, T.; et al. Assessing Cellular Uptake of Exogenous Coenzyme Q(10) into Human Skin Cells by X-ray Fluorescence Imaging. Antioxidants 2022, 11, 1532. [Google Scholar] [CrossRef]
- Kleiner, G.; Barca, E.; Ziosi, M.; Emmanuele, V.; Xu, Y.; Hidalgo-Gutierrez, A.; Qiao, C.; Tadesse, S.; Area-Gomez, E.; Lopez, L.C.; et al. CoQ(10) supplementation rescues nephrotic syndrome through normalization of H(2)S oxidation pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3708–3722. [Google Scholar] [CrossRef]
- Wang, Y.; Hekimi, S. Micellization of coenzyme Q by the fungicide caspofungin allows for safe intravenous administration to reach extreme supraphysiological concentrations. Redox Biol. 2020, 36, 101680. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gumus, E.; Hekimi, S. A novel COQ7 mutation causing primarily neuromuscular pathology and its treatment options. Mol. Genet. Metab. Rep. 2022, 31, 100877. [Google Scholar] [CrossRef]
- Wainwright, L.; Hargreaves, I.P.; Georgian, A.R.; Turner, C.; Dalton, R.N.; Abbott, N.J.; Heales, S.J.R.; Preston, J.E. CoQ(10) Deficient Endothelial Cell Culture Model for the Investigation of CoQ(10) Blood-Brain Barrier Transport. J. Clin. Med. 2020, 9, 3236. [Google Scholar] [CrossRef]
- Balazs, Z.; Panzenboeck, U.; Hammer, A.; Sovic, A.; Quehenberger, O.; Malle, E.; Sattler, W. Uptake and transport of high-density lipoprotein (HDL) and HDL-associated alpha-tocopherol by an in vitro blood-brain barrier model. J. Neurochem. 2004, 89, 939–950. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, D.; Zhao, J.; Song, J.; Zhao, Y. The role of the low-density lipoprotein receptor-related protein 1 (LRP-1) in regulating blood-brain barrier integrity. Rev. Neurosci. 2016, 27, 623–634. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Zlokovic, B.V. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 2004, 35, 2628–2631. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Itagaki, S.; Ochiai, A.; Kobayashi, M.; Sugawara, M.; Hirano, T.; Iseki, K. Interaction of coenzyme Q10 with the intestinal drug transporter P-glycoprotein. J. Agric. Food Chem. 2008, 56, 6923–6927. [Google Scholar] [CrossRef] [PubMed]
- Powers, H.R.; Sahoo, D. SR-B1′s Next Top Model: Structural Perspectives on the Functions of the HDL Receptor. Curr. Atheroscler. Rep. 2022, 24, 277–288. [Google Scholar] [CrossRef]
- Li, Y.; Huang, X.; Yang, G.; Xu, K.; Yin, Y.; Brecchia, G.; Yin, J. CD36 favours fat sensing and transport to govern lipid metabolism. Prog. Lipid Res. 2022, 88, 101193. [Google Scholar] [CrossRef]
- Anderson, C.M.; Kazantzis, M.; Wang, J.; Venkatraman, S.; Goncalves, R.L.; Quinlan, C.L.; Ng, R.; Jastroch, M.; Benjamin, D.I.; Nie, B.; et al. Dependence of brown adipose tissue function on CD36-mediated coenzyme Q uptake. Cell Rep. 2015, 10, 505–515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kishimoto, Y.; Hiraiwa, M.; O’Brien, J.S. Saposins: Structure, function, distribution, and molecular genetics. J. Lipid Res. 1992, 33, 1255–1267. [Google Scholar]
- Ciaffoni, F.; Tatti, M.; Boe, A.; Salvioli, R.; Fluharty, A.; Sonnino, S.; Vaccaro, A.M. Saposin B binds and transfers phospholipids. J. Lipid Res. 2006, 47, 1045–1053. [Google Scholar] [CrossRef][Green Version]
- Ahn, V.E.; Faull, K.F.; Whitelegge, J.P.; Fluharty, A.L.; Prive, G.G. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc. Natl. Acad. Sci. USA 2003, 100, 38–43. [Google Scholar] [CrossRef]
- Dixson, D.D.; Yu, V.Y.; Doyle, R.P. Recombinant expression of His-tagged saposin B and pH-dependent binding to the lipid coenzyme Q10. Anal. Biochem. 2011, 419, 145–152. [Google Scholar] [CrossRef]
- Huta, B.P.; Mehlenbacher, M.R.; Nie, Y.; Lai, X.; Zubieta, C.; Bou-Abdallah, F.; Doyle, R.P. The Lysosomal Protein Saposin B Binds Chloroquine. ChemMedChem 2016, 11, 277–282. [Google Scholar] [CrossRef][Green Version]
- Tinklepaugh, J.; Smith, B.M.; Hanlon, E.; Zubieta, C.; Bou-Abdallah, F.; Doyle, R.P. Exploring the Multiligand Binding Specificity of Saposin B Reveals Two Binding Sites. ACS Omega 2017, 2, 7141–7145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kashiba, M.; Oizumi, M.; Suzuki, M.; Sawamura, Y.; Nagashima, K.; Yoshimura, S.; Yamamoto, Y. Prosaposin regulates coenzyme Q10 levels in HepG2 cells, especially those in mitochondria. J. Clin. Biochem. Nutr. 2014, 55, 85–89. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kashiba, M.; Terashima, M.; Sagawa, T.; Yoshimura, S.; Yamamoto, Y. Prosaposin knockdown in Caco-2 cells decreases cellular levels of coenzyme Q10 and ATP, and results in the loss of tight junction barriers. J. Clin. Biochem. Nutr. 2017, 60, 81–85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takeuchi, H.; Sugawara, K.; Okamoto, M.; Nakamura, A.; Tanaka, T.; Fujita, Y.; Ishiguro, K.; Yamazaki, H.; Okada, M.; Mikami, A.; et al. Reduced prosaposin levels in HepG2 cells with long-term coenzyme Q10 deficiency. J. Clin. Biochem. Nutr. 2022, 71, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Suzuki, K.; Vanier, M.T.; Popko, B.; Maeda, N.; Klein, A.; Henseler, M.; Sandhoff, K.; Nakayasu, H.; Suzuki, K. Targeted disruption of the mouse sphingolipid activator protein gene: A complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum. Mol. Genet. 1996, 5, 711–725. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, P.; Barriocanal-Casado, E.; Diaz-Casado, M.E.; Lopez-Herrador, S.; Hidalgo-Gutierrez, A.; Lopez, L.C. Animal Models of Coenzyme Q Deficiency: Mechanistic and Translational Learnings. Antioxidants 2021, 10, 1687. [Google Scholar] [CrossRef]
- Licitra, F.; Puccio, H. An overview of current mouse models recapitulating coenzyme q10 deficiency syndrome. Mol. Syndromol. 2014, 5, 180–186. [Google Scholar] [CrossRef][Green Version]
- Raizner, A.E. Coenzyme Q(10). Methodist Debakey Cardiovasc. J. 2019, 15, 185–191. [Google Scholar] [CrossRef]
- Bhagavan, H.N.; Chopra, R.K. Coenzyme Q10: Absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic. Res. 2006, 40, 445–453. [Google Scholar] [CrossRef]
- Sohal, R.S.; Kamzalov, S.; Sumien, N.; Ferguson, M.; Rebrin, I.; Heinrich, K.R.; Forster, M.J. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic. Biol. Med. 2006, 40, 480–487. [Google Scholar] [CrossRef][Green Version]
- Palamakula, A.; Soliman, M.; Khan, M.M. Regional permeability of coenzyme Q10 in isolated rat gastrointestinal tracts. Pharmazie 2005, 60, 212–214. [Google Scholar]
- Hokkanen, K.; Tirronen, A.; Yla-Herttuala, S. Intestinal lymphatic vessels and their role in chylomicron absorption and lipid homeostasis. Curr. Opin. Lipidol. 2019, 30, 370–376. [Google Scholar] [CrossRef]
- Katayama, K.; Fujita, T. Studies on lymphatic absorption of 1′,2′-(3 H)-coenzyme Q 10 in rats. Chem. Pharm. Bull. 1972, 20, 2585–2592. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kono, N.; Arai, H. Intracellular transport of fat-soluble vitamins A and E. Traffic 2015, 16, 19–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feingold, K.R. Introduction to Lipids and Lipoproteins. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Tomasetti, M.; Alleva, R.; Solenghi, M.D.; Littarru, G.P. Distribution of antioxidants among blood components and lipoproteins: Significance of lipids/CoQ10 ratio as a possible marker of increased risk for atherosclerosis. BioFactors 1999, 9, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Mohr, D.; Bowry, V.W.; Stocker, R. Dietary supplementation with coenzyme Q10 results in increased levels of ubiquinol-10 within circulating lipoproteins and increased resistance of human low-density lipoprotein to the initiation of lipid peroxidation. Biochim. Biophys. Acta 1992, 1126, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Bowry, V.W.; Frei, B. Ubiquinol-10 protects human low density lipoprotein more efficiently against lipid peroxidation than does alpha-tocopherol. Proc. Natl. Acad. Sci. USA 1991, 88, 1646–1650. [Google Scholar] [CrossRef]
- Dadabayev, A.R.; Yin, G.; Latchoumycandane, C.; McIntyre, T.M.; Lesnefsky, E.J.; Penn, M.S. Apolipoprotein A1 regulates coenzyme Q10 absorption, mitochondrial function, and infarct size in a mouse model of myocardial infarction. J. Nutr. 2014, 144, 1030–1036. [Google Scholar] [CrossRef][Green Version]
- Lonnrot, K.; Holm, P.; Lagerstedt, A.; Huhtala, H.; Alho, H. The effects of lifelong ubiquinone Q10 supplementation on the Q9 and Q10 tissue concentrations and life span of male rats and mice. Biochem. Mol. Biol. Int. 1998, 44, 727–737. [Google Scholar] [CrossRef]
- Kwong, L.K.; Kamzalov, S.; Rebrin, I.; Bayne, A.C.; Jana, C.K.; Morris, P.; Forster, M.J.; Sohal, R.S. Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free. Radic. Biol. Med. 2002, 33, 627–638. [Google Scholar] [CrossRef]
- Lass, A.; Forster, M.J.; Sohal, R.S. Effects of coenzyme Q10 and alpha-tocopherol administration on their tissue levels in the mouse: Elevation of mitochondrial alpha-tocopherol by coenzyme Q10. Free. Radic. Biol. Med. 1999, 26, 1375–1382. [Google Scholar] [CrossRef]
- Kamzalov, S.; Sumien, N.; Forster, M.J.; Sohal, R.S. Coenzyme Q intake elevates the mitochondrial and tissue levels of Coenzyme Q and alpha-tocopherol in young mice. J. Nutr. 2003, 133, 3175–3180. [Google Scholar] [CrossRef][Green Version]
- Lekli, I.; Das, S.; Das, S.; Mukherjee, S.; Bak, I.; Juhasz, B.; Bagchi, D.; Trimurtulu, G.; Krishnaraju, A.V.; Sengupta, K.; et al. Coenzyme Q9 provides cardioprotection after converting into coenzyme Q10. J. Agric. Food Chem. 2008, 56, 5331–5337. [Google Scholar] [CrossRef] [PubMed]
- Argyropoulos, G.; Harper, M.E. Uncoupling proteins and thermoregulation. J. Appl. Physiol. 2002, 92, 2187–2198. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martelli, A.; Testai, L.; Colletti, A.; Cicero, A.F.G. Coenzyme Q(10): Clinical Applications in Cardiovascular Diseases. Antioxidants 2020, 9, 341. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garcia-Corzo, L.; Luna-Sanchez, M.; Doerrier, C.; Garcia, J.A.; Guaras, A.; Acin-Perez, R.; Bullejos-Peregrin, J.; Lopez, A.; Escames, G.; Enriquez, J.A.; et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2013, 22, 1233–1248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Watanabe, K.; Nozaki, S.; Goto, M.; Kaneko, K.I.; Hayashinaka, E.; Irie, S.; Nishiyama, A.; Kasai, K.; Fujii, K.; Wada, Y.; et al. PET imaging of (11)C-labeled coenzyme Q(10): Comparison of biodistribution between [(11)C]ubiquinol-10 and [(11)C]ubiquinone-10. Biochem. Biophys. Res. Commun. 2019, 512, 611–615. [Google Scholar] [CrossRef]
- Bentinger, M.; Dallner, G.; Chojnacki, T.; Swiezewska, E. Distribution and breakdown of labeled coenzyme Q10 in rat. Free Radic. Biol. Med. 2003, 34, 563–575. [Google Scholar] [CrossRef]
- Reahal, S.; Wrigglesworth, J. Tissue concentrations of coenzyme Q10 in the rat following its oral and intraperitoneal administration. Drug Metab. Dispos. 1992, 20, 423–427. [Google Scholar]
- Scalori, V.; Alessandri, M.G.; Giovannini, L.; Bertelli, A. Plasma and tissue concentrations of coenzyme Q10 in the rat after intravenous, oral and topical administrations. Int. J. Tissue React. 1990, 12, 149–154. [Google Scholar]
- Alessandri, M.G.; Scalori, V.; Giovannini, L.; Mian, M.; Bertelli, A.A. Plasma and tissue concentrations of coenzyme Q10 in the rat after intravenous administration by a microsphere delivery system or in a new type of solution. Int. J. Tissue React. 1988, 10, 99–102. [Google Scholar] [PubMed]
- Knott, A.; Achterberg, V.; Smuda, C.; Mielke, H.; Sperling, G.; Dunckelmann, K.; Vogelsang, A.; Kruger, A.; Schwengler, H.; Behtash, M.; et al. Topical treatment with coenzyme Q10-containing formulas improves skin’s Q10 level and provides antioxidative effects. BioFactors 2015, 41, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Ooe, M.; Fujii, K.; Matsunaka, H.; Yoshida, M.; Ichihashi, M. Mechanisms of inhibitory effects of CoQ10 on UVB-induced wrinkle formation in vitro and in vivo. BioFactors 2008, 32, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, Y.; Kasai, K.; Maruyama, C.; Hamano, Y.; Matsuo, K.; Katano, H.; Taira, S. Imaging mass spectrometry analysis of ubiquinol localization in the mouse brain following short-term administration. Sci. Rep. 2017, 7, 12990. [Google Scholar] [CrossRef][Green Version]
- Passi, S.; Maggio, F.; Ricci, R.; Cocchi, M. Prolonged administration of ubiquinone (CoQ10) and ubiquinol diacetate (CoQ10 DIA) to rats and guinea pigs. Prog. Nutr. 2003, 5, 6–15. [Google Scholar]
- Zhang, Y.; Aberg, F.; Appelkvist, E.L.; Dallner, G.; Ernster, L. Uptake of dietary coenzyme Q supplement is limited in rats. J. Nutr. 1995, 125, 446–453. [Google Scholar] [CrossRef]
- Saiki, R.; Lunceford, A.L.; Shi, Y.; Marbois, B.; King, R.; Pachuski, J.; Kawamukai, M.; Gasser, D.L.; Clarke, C.F. Coenzyme Q10 supplementation rescues renal disease in Pdss2kd/kd mice with mutations in prenyl diphosphate synthase subunit 2. Am. J. Physiol. Renal Physiol. 2008, 295, F1535–F1544. [Google Scholar] [CrossRef][Green Version]
- Peng, M.; Falk, M.J.; Haase, V.H.; King, R.; Polyak, E.; Selak, M.; Yudkoff, M.; Hancock, W.W.; Meade, R.; Saiki, R.; et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008, 4, e1000061. [Google Scholar] [CrossRef][Green Version]
- Tryggvason, K.; Patrakka, J.; Wartiovaara, J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N. Engl. J. Med. 2006, 354, 1387–1401. [Google Scholar] [CrossRef][Green Version]
- Ziosi, M.; Di Meo, I.; Kleiner, G.; Gao, X.H.; Barca, E.; Sanchez-Quintero, M.J.; Tadesse, S.; Jiang, H.; Qiao, C.; Rodenburg, R.J.; et al. Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol. Med. 2017, 9, 96–111. [Google Scholar] [CrossRef]
- Luna-Sanchez, M.; Hidalgo-Gutierrez, A.; Hildebrandt, T.M.; Chaves-Serrano, J.; Barriocanal-Casado, E.; Santos-Fandila, A.; Romero, M.; Sayed, R.K.; Duarte, J.; Prokisch, H.; et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol. Med. 2017, 9, 78–95. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, P.; Hidalgo-Gutierrez, A.; Mascaraque, C.; Barriocanal-Casado, E.; Bakkali, M.; Ziosi, M.; Abdihankyzy, U.B.; Sanchez-Hernandez, S.; Escames, G.; Prokisch, H.; et al. Coenzyme Q10 modulates sulfide metabolism and links the mitochondrial respiratory chain to pathways associated to one carbon metabolism. Hum. Mol. Genet. 2020, 29, 3296–3311. [Google Scholar] [CrossRef]
- Lapointe, J.; Wang, Y.; Bigras, E.; Hekimi, S. The submitochondrial distribution of ubiquinone affects respiration in long-lived Mclk1+/− mice. J. Cell Biol. 2012, 199, 215–224. [Google Scholar] [CrossRef][Green Version]
- Lapointe, J.; Hekimi, S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J. Biol. Chem. 2008, 283, 26217–26227. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lapointe, J.; Stepanyan, Z.; Bigras, E.; Hekimi, S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/− mice. J. Biol. Chem. 2009, 284, 20364–20374. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alcazar-Fabra, M.; Rodriguez-Sanchez, F.; Trevisson, E.; Brea-Calvo, G. Primary Coenzyme Q deficiencies: A literature review and online platform of clinical features to uncover genotype-phenotype correlations. Free Radic. Biol. Med. 2021, 167, 141–180. [Google Scholar] [CrossRef] [PubMed]
- Brea-Calvo, G.; Haack, T.B.; Karall, D.; Ohtake, A.; Invernizzi, F.; Carrozzo, R.; Kremer, L.; Dusi, S.; Fauth, C.; Scholl-Burgi, S.; et al. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am. J. Hum. Genet. 2015, 96, 309–317. [Google Scholar] [CrossRef][Green Version]
- Salviati, L.; Sacconi, S.; Murer, L.; Zacchello, G.; Franceschini, L.; Laverda, A.M.; Basso, G.; Quinzii, C.; Angelini, C.; Hirano, M.; et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: A CoQ10-responsive condition. Neurology 2005, 65, 606–608. [Google Scholar] [CrossRef]
- Quinzii, C.M.; Hirano, M.; DiMauro, S. CoQ10 deficiency diseases in adults. Mitochondrion 2007, 7, S122–S126. [Google Scholar] [CrossRef][Green Version]
- Potgieter, M.; Pretorius, E.; Pepper, M.S. Primary and secondary coenzyme Q10 deficiency: The role of therapeutic supplementation. Nutr. Rev. 2013, 71, 180–188. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Coenzyme Q functions. (a) Mitochondrial CoQ shuttles electrons in the electron transport chain and is reduced by dehydrogenases in diverse metabolic pathways. CI, Complex I; CII, Complex II; CIII, Complex III; CIV, Complex IV; CV, Complex V; DHODH, dihydroorotate dehydrogenase; GPDH, glyceraldehyde-3-phosphate dehydrogenase; SQR, sulfide:quinone oxidoreductase; CHDH, choline dehydrogenase; PRDH, proline dehydrogenase; ETF, electron transfer flavoprotein; ETFDH, electron transfer flavoprotein dehydrogenase. (b) CoQH2 is present in cellular membranes and lipoproteins and acts as a chain-terminating antioxidant to inhibit both the initiation and propagation steps of lipid autoxidation. CoQH2 or ascorbate regenerate vitamin E from the tocopheroxyl radical. (c) CoQ functions in the plasma membrane redox system (PMRS) with other antioxidants, such as vitamin E and ascorbate. CoQ is reduced by NAD(P)H:quinone oxidoreductase 1 (NQO1), or NADH-cytochrome b5 reductase (cyb5Red). (d) The CoQ reductase ferroptosis suppressor protein 1 (FSP1) regenerates CoQH2 at the plasma membrane to terminate lipid peroxidation and suppress ferroptosis. Created with Biorender.com (accessed on 23 May 2023).
Figure 1.
Coenzyme Q functions. (a) Mitochondrial CoQ shuttles electrons in the electron transport chain and is reduced by dehydrogenases in diverse metabolic pathways. CI, Complex I; CII, Complex II; CIII, Complex III; CIV, Complex IV; CV, Complex V; DHODH, dihydroorotate dehydrogenase; GPDH, glyceraldehyde-3-phosphate dehydrogenase; SQR, sulfide:quinone oxidoreductase; CHDH, choline dehydrogenase; PRDH, proline dehydrogenase; ETF, electron transfer flavoprotein; ETFDH, electron transfer flavoprotein dehydrogenase. (b) CoQH2 is present in cellular membranes and lipoproteins and acts as a chain-terminating antioxidant to inhibit both the initiation and propagation steps of lipid autoxidation. CoQH2 or ascorbate regenerate vitamin E from the tocopheroxyl radical. (c) CoQ functions in the plasma membrane redox system (PMRS) with other antioxidants, such as vitamin E and ascorbate. CoQ is reduced by NAD(P)H:quinone oxidoreductase 1 (NQO1), or NADH-cytochrome b5 reductase (cyb5Red). (d) The CoQ reductase ferroptosis suppressor protein 1 (FSP1) regenerates CoQH2 at the plasma membrane to terminate lipid peroxidation and suppress ferroptosis. Created with Biorender.com (accessed on 23 May 2023).

Figure 3.
Yeast coq mutants are rescued with exogenous CoQ6 but not non-isoprenoid CoQ analogues. (a) CoQ6 was not detected in any of the coqΔ mutants. CoQ6 (pmol/mg protein) was determined based on a CoQ6 standard curve as described in (Tsui et al., 2019). (b) Each of the coq mutants, from coq3Δ to coq9Δ, accumulated early CoQ6 intermediates (HHB and HAB). The amount of HHB and HAB was calculated as the area of the MS peak/mg protein. (c) Yeast mutants harboring deletions in either the COQ1 or COQ2 gene showed more robust rescue in response to exogenous CoQ6 treatment than do the other single coq null mutants (coq3Δ–coq9Δ). (d) Additional deletions of either COQ1 or COQ2 restored the deficient CoQ6-rescue of a coq3Δ mutant, but do not affect the phenotype observed in coq1Δ or coqΔ2 strains. Columns represent the degree of rescue (in %) ± SD of a strain compared to WT, which is defined as 100% and represented as a dashed line. Statistically significant differences between a specific coqΔ mutant and one of its counterparts (another coqΔ mutant) are denoted with numbers in parentheses on top of the columns. (1) represents differences comparing to coq1Δ, (2) represents differences comparing to coq2Δ, (3) represents differences comparing to coq3Δ, etc. Three or more independent rescue experiments were performed for every strain. Asterisks on top of the columns represent significant differences when compared to WT (* p < 0.05, ** p < 0.01, *** p < 0.001). (e) Structures of CoQ analogues. Panels (a–d) reproduced with permission from Fernandez-del-Rio et al. Free Radicals in Biology and Medicine, published by Elsevier, 2020 [18]. Created with Biorender.com (Accessed on 27 June 2023).
Figure 3.
Yeast coq mutants are rescued with exogenous CoQ6 but not non-isoprenoid CoQ analogues. (a) CoQ6 was not detected in any of the coqΔ mutants. CoQ6 (pmol/mg protein) was determined based on a CoQ6 standard curve as described in (Tsui et al., 2019). (b) Each of the coq mutants, from coq3Δ to coq9Δ, accumulated early CoQ6 intermediates (HHB and HAB). The amount of HHB and HAB was calculated as the area of the MS peak/mg protein. (c) Yeast mutants harboring deletions in either the COQ1 or COQ2 gene showed more robust rescue in response to exogenous CoQ6 treatment than do the other single coq null mutants (coq3Δ–coq9Δ). (d) Additional deletions of either COQ1 or COQ2 restored the deficient CoQ6-rescue of a coq3Δ mutant, but do not affect the phenotype observed in coq1Δ or coqΔ2 strains. Columns represent the degree of rescue (in %) ± SD of a strain compared to WT, which is defined as 100% and represented as a dashed line. Statistically significant differences between a specific coqΔ mutant and one of its counterparts (another coqΔ mutant) are denoted with numbers in parentheses on top of the columns. (1) represents differences comparing to coq1Δ, (2) represents differences comparing to coq2Δ, (3) represents differences comparing to coq3Δ, etc. Three or more independent rescue experiments were performed for every strain. Asterisks on top of the columns represent significant differences when compared to WT (* p < 0.05, ** p < 0.01, *** p < 0.001). (e) Structures of CoQ analogues. Panels (a–d) reproduced with permission from Fernandez-del-Rio et al. Free Radicals in Biology and Medicine, published by Elsevier, 2020 [18]. Created with Biorender.com (Accessed on 27 June 2023).

Figure 4.
Pathways and proteins involved in trafficking of CoQ6 to and from mitochondria. See text for the description of gene products that are involved in transport of CoQ6 in yeast. The endoplasmic reticulum-mitochondria encounter structure (ERMES) is formed by Mdm10 and Mdm34 at the outer mitochondrial membrane, Mdm12 in the cytosol, and Mmm1 on the ER membrane. Septin filaments are hetero-oligomeric complexes composed of septin subunits, including Cdc10, Cdc3, Cdc12, Cdc11, and sometimes Shs1. PP2A, protein phosphatase 2A. Dashed lines denoted with question marks indicate postulated pathways of CoQ trafficking. Created with Biorender.com (Accessed on 25 May 2023).
Figure 4.
Pathways and proteins involved in trafficking of CoQ6 to and from mitochondria. See text for the description of gene products that are involved in transport of CoQ6 in yeast. The endoplasmic reticulum-mitochondria encounter structure (ERMES) is formed by Mdm10 and Mdm34 at the outer mitochondrial membrane, Mdm12 in the cytosol, and Mmm1 on the ER membrane. Septin filaments are hetero-oligomeric complexes composed of septin subunits, including Cdc10, Cdc3, Cdc12, Cdc11, and sometimes Shs1. PP2A, protein phosphatase 2A. Dashed lines denoted with question marks indicate postulated pathways of CoQ trafficking. Created with Biorender.com (Accessed on 25 May 2023).

Figure 5.
CaCo-2 cell monolayers are used to model the uptake and transport of CoQ from the apical or lumenal side to the basolateral compartment. Created with BioRender.com (Accessed on 25 May 2023).
Figure 5.
CaCo-2 cell monolayers are used to model the uptake and transport of CoQ from the apical or lumenal side to the basolateral compartment. Created with BioRender.com (Accessed on 25 May 2023).

Figure 6.
CoQ10 uptake and trafficking in mammalian cells. Dashed arrows indicate CoQ movement; solid arrows indicate chemical reactions. Proteins responsible for trafficking CoQ are identified where known; proteins mediating the transport shown by dashed arrows remain to be identified. (a) Exogenous CoQ uptake and intracellular CoQ distribution are mediated by several known mechanisms in mammalian cell culture. CD36 and NPC1L1 recognize and import exogenous CoQ within brown adipose tissue and small intestine epithelial cells, respectively. The endomembrane system facilitates intracellular trafficking of CoQ between membrane-bound vesicles. Dual-localized STARD7 in the mitochondria and cytosol support endogenous CoQ biosynthesis and distribution to the plasma membrane, respectively. (b) Model of lipoprotein-associated CoQ trafficking at the blood–brain barrier. SR-B1 and RAGE regulate influx of the HDL-associated CoQ across the blood–brain barrier, while LRP-1 regulates the efflux of LDL-associated CoQ. 1, receptor-mediated endocytosis; 2, recycling to apical side via the same receptor; 3, transcytosis to basolateral side; 4, transport to lysosome; 5, recycling or efflux of CoQ to apical membrane via LDL receptor family proteins; 6, paracellular transport detected due to defects in tight junctions. HDL, high-density lipoprotein; LDL, low-density lipoprotein. Created with BioRender.com (Accessed on 28 June 2023).
Figure 6.
CoQ10 uptake and trafficking in mammalian cells. Dashed arrows indicate CoQ movement; solid arrows indicate chemical reactions. Proteins responsible for trafficking CoQ are identified where known; proteins mediating the transport shown by dashed arrows remain to be identified. (a) Exogenous CoQ uptake and intracellular CoQ distribution are mediated by several known mechanisms in mammalian cell culture. CD36 and NPC1L1 recognize and import exogenous CoQ within brown adipose tissue and small intestine epithelial cells, respectively. The endomembrane system facilitates intracellular trafficking of CoQ between membrane-bound vesicles. Dual-localized STARD7 in the mitochondria and cytosol support endogenous CoQ biosynthesis and distribution to the plasma membrane, respectively. (b) Model of lipoprotein-associated CoQ trafficking at the blood–brain barrier. SR-B1 and RAGE regulate influx of the HDL-associated CoQ across the blood–brain barrier, while LRP-1 regulates the efflux of LDL-associated CoQ. 1, receptor-mediated endocytosis; 2, recycling to apical side via the same receptor; 3, transcytosis to basolateral side; 4, transport to lysosome; 5, recycling or efflux of CoQ to apical membrane via LDL receptor family proteins; 6, paracellular transport detected due to defects in tight junctions. HDL, high-density lipoprotein; LDL, low-density lipoprotein. Created with BioRender.com (Accessed on 28 June 2023).

Figure 7.
Model for the absorption of dietary-supplemented CoQ in mice. Dashed arrows indicate CoQ distribution to tissues enriched only at high doses or IP injection. Stars in urine and feces indicate metabolites of CoQ. The ? indicates an unknown mechanism for the uptake of CoQ into the brain. VLDL—very low-density lipoprotein; IDL—intermediate-density lipoprotein; LDL—low-density lipoprotein; LPL—lipoprotein lipase. Created with BioRender.com (Accessed 31 January 2023).
Figure 7.
Model for the absorption of dietary-supplemented CoQ in mice. Dashed arrows indicate CoQ distribution to tissues enriched only at high doses or IP injection. Stars in urine and feces indicate metabolites of CoQ. The ? indicates an unknown mechanism for the uptake of CoQ into the brain. VLDL—very low-density lipoprotein; IDL—intermediate-density lipoprotein; LDL—low-density lipoprotein; LPL—lipoprotein lipase. Created with BioRender.com (Accessed 31 January 2023).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Guile, M.D.; Jain, A.; Anderson, K.A.; Clarke, C.F. New Insights on the Uptake and Trafficking of Coenzyme Q. Antioxidants 2023, 12, 1391. https://doi.org/10.3390/antiox12071391
AMA Style
Guile MD, Jain A, Anderson KA, Clarke CF. New Insights on the Uptake and Trafficking of Coenzyme Q. Antioxidants. 2023; 12(7):1391. https://doi.org/10.3390/antiox12071391
Chicago/Turabian StyleGuile, Michael D., Akash Jain, Kyle A. Anderson, and Catherine F. Clarke. 2023. "New Insights on the Uptake and Trafficking of Coenzyme Q" Antioxidants 12, no. 7: 1391. https://doi.org/10.3390/antiox12071391
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.