


by

Epigenomes 2023, 7(3), 19; https://doi.org/10.3390/epigenomes7030019 - 22 Aug 2023
Abstract
►
Show Figures
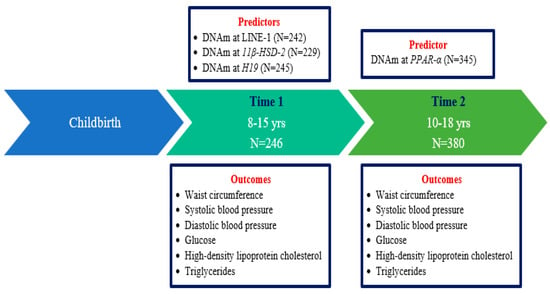
Understanding the epigenome paths through which smoking contributes to cardiometabolic traits is important for downstream applications. In this study, an SNP-based analytical pipeline was used to integrate several publicly available datasets in order to identify CpG sites that mediate the impact of smoking
[...] Read more.
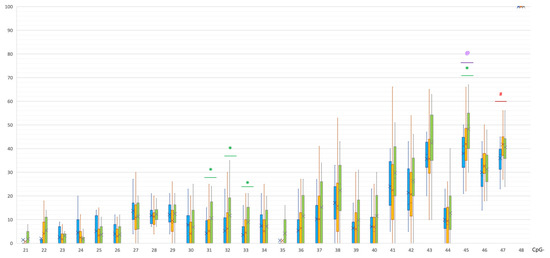
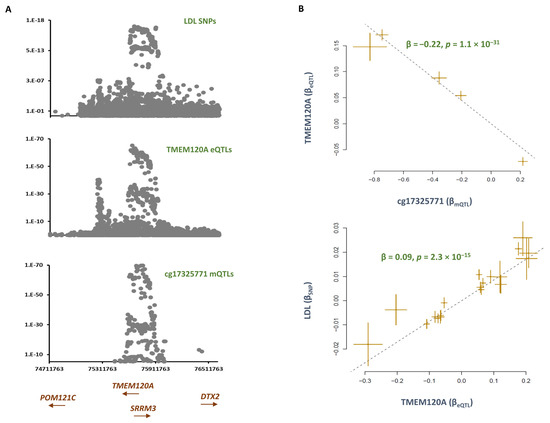
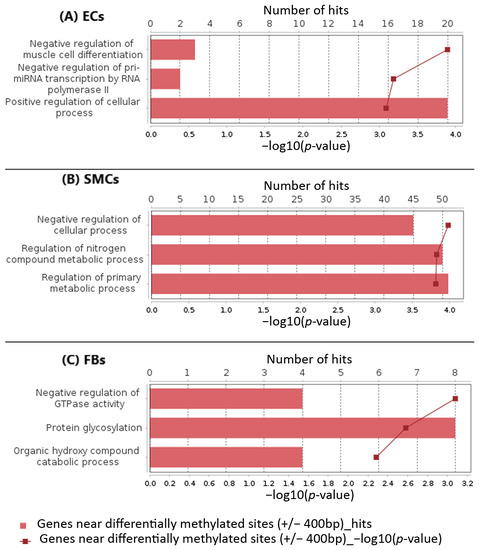
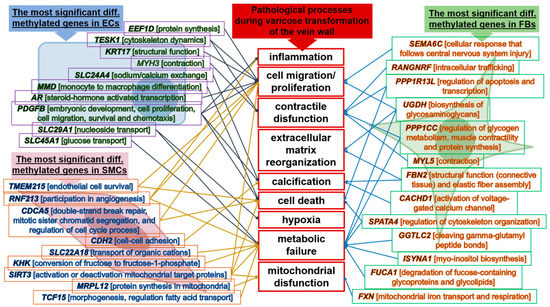
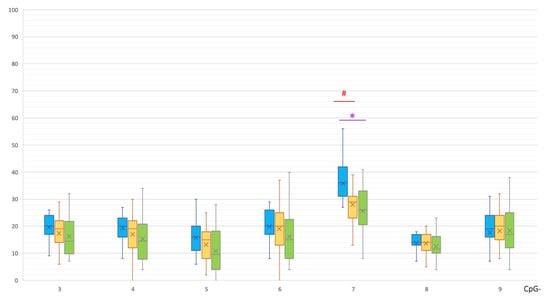
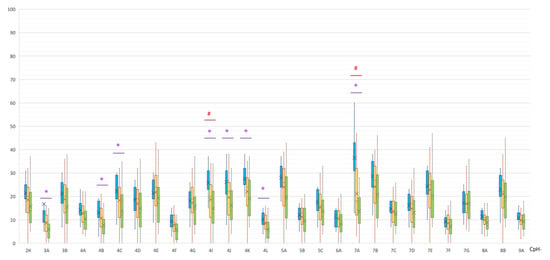
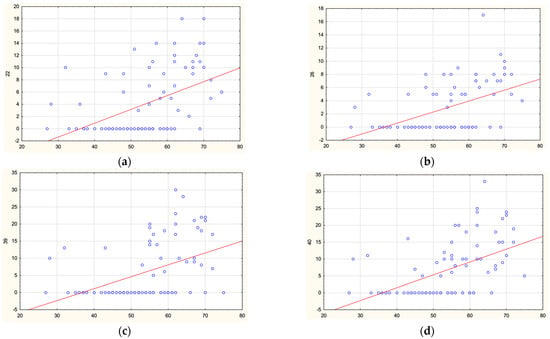
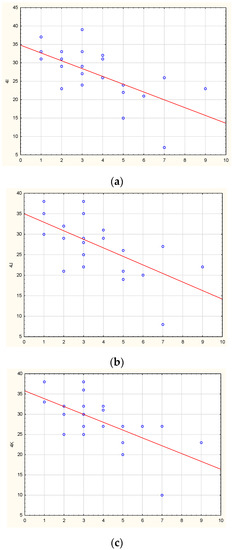
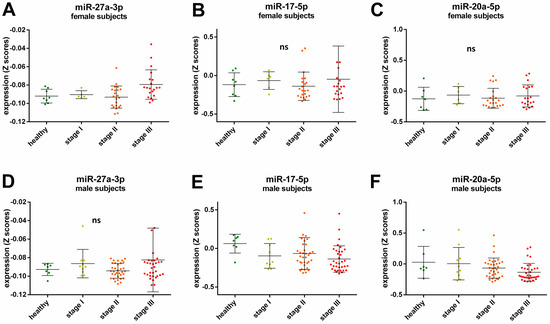
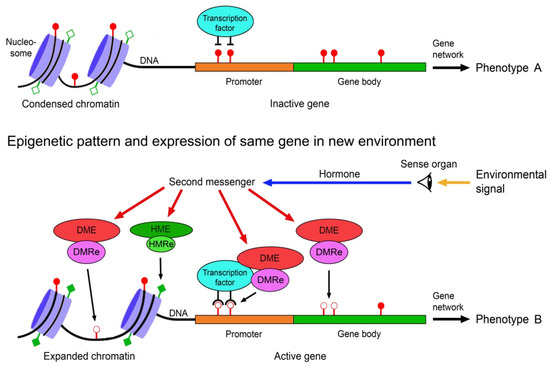
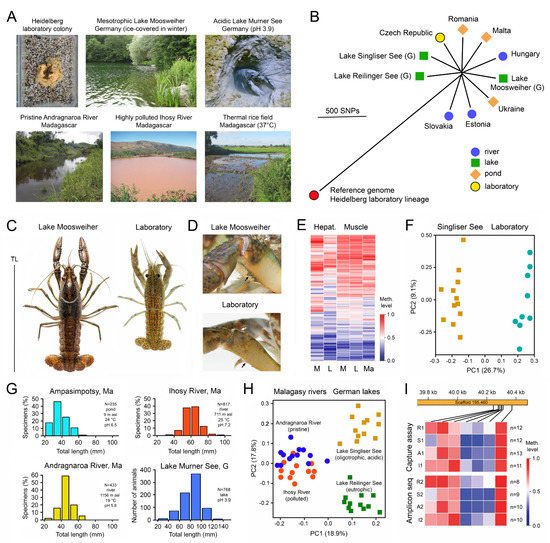
Understanding the epigenome paths through which smoking contributes to cardiometabolic traits is important for downstream applications. In this study, an SNP-based analytical pipeline was used to integrate several publicly available datasets in order to identify CpG sites that mediate the impact of smoking on cardiometabolic traits and to investigate the underlying molecular mechanisms. After applying stringent statistical criteria, 11 CpG sites were detected that showed significant association (p < 5 × 10−8) with cardiometabolic traits at both the discovery and replication stages. By integrating eQTL data, I found genes behind a number of these associations. cg05228408 was hypomethylated in smokers and contributed to higher blood pressure by lowering the expression of the CLCN6 gene. cg08639339 was hypermethylated in smokers and lowered the metabolic rate by increasing the expression of RAB29; furthermore, I noted TMEM120A mediated the impact of smoking-cg17325771 on LDL, and LTBP3 mediated the smoking-cg07029024 effect on heart rate. The pathway analysis identified processes through which the identified genes impact their traits. This study provides a list of CpG sites that mediates the impact of smoking on cardiometabolic traits and a framework to investigate the underlying molecular paths using publicly available data.
Full article

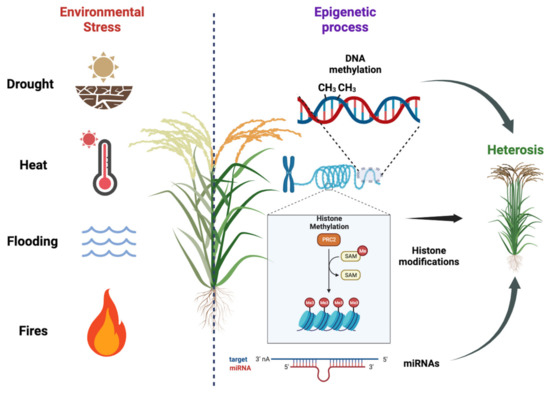
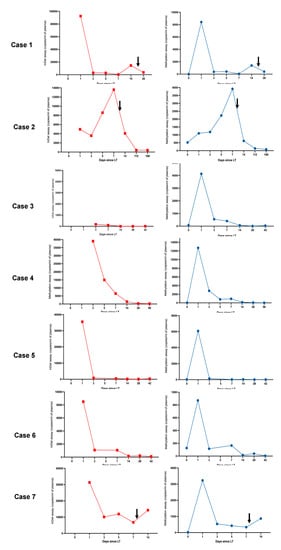
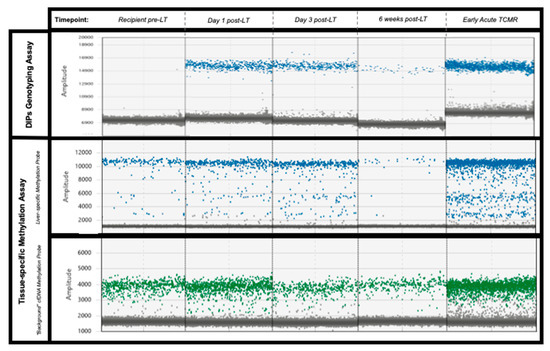
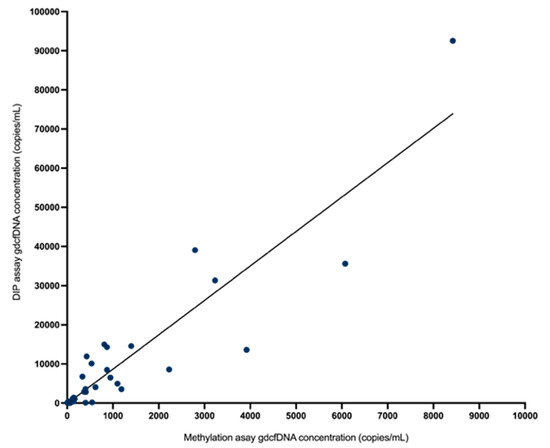
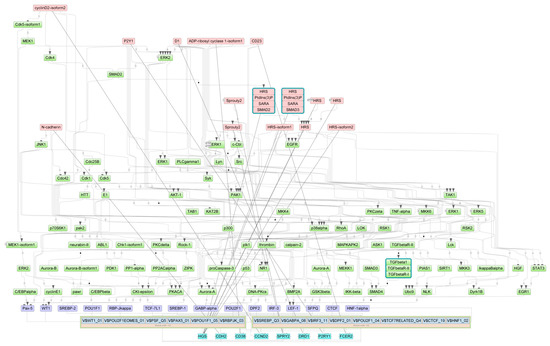
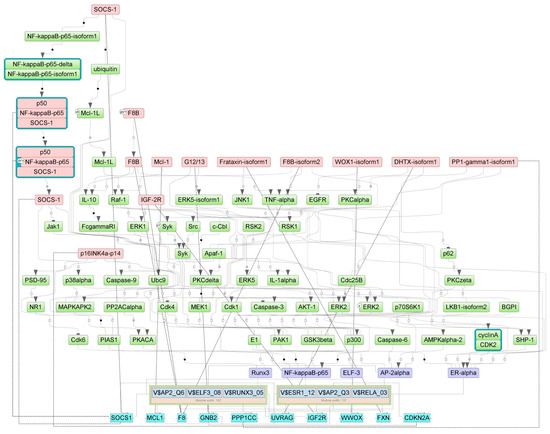
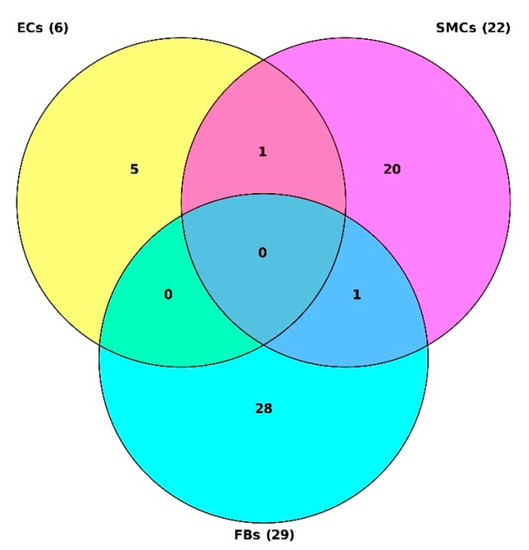
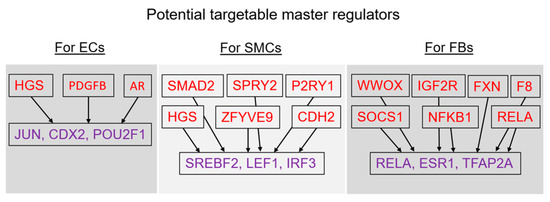
Figure 1















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}