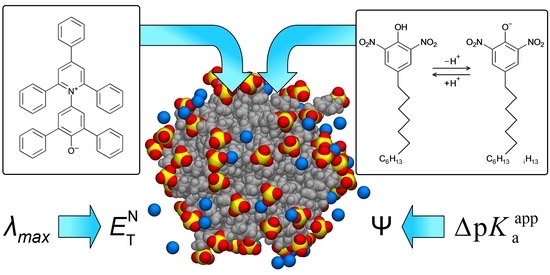
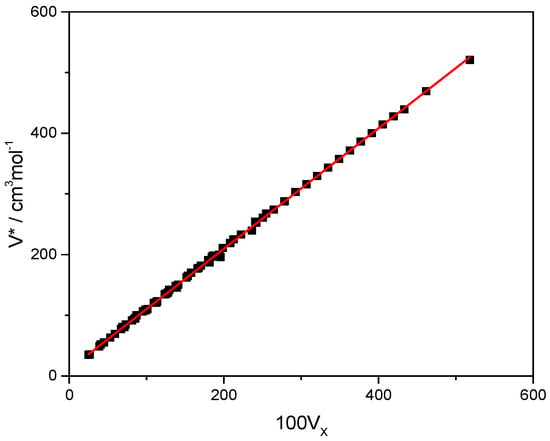
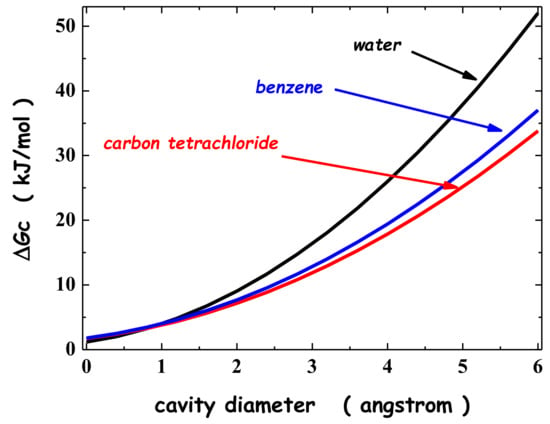
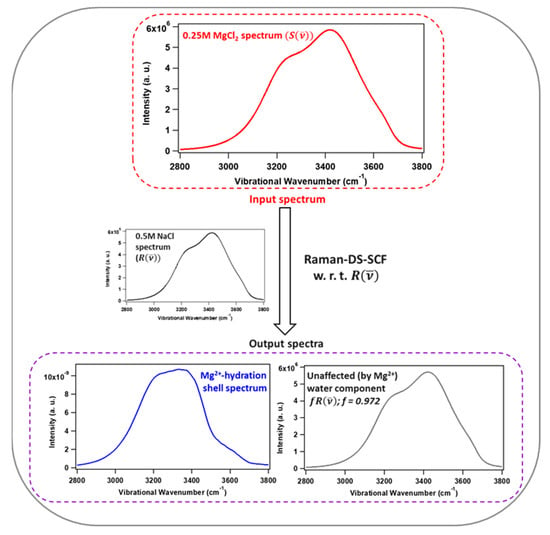
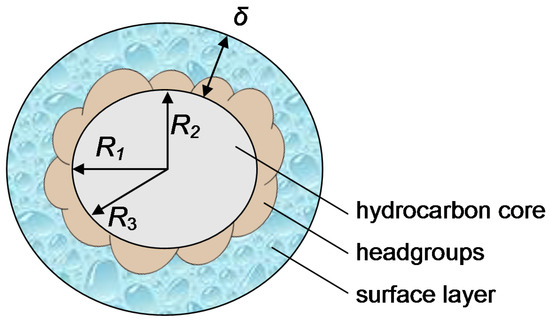
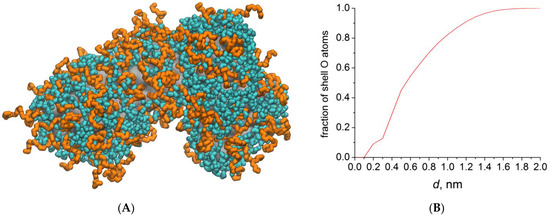
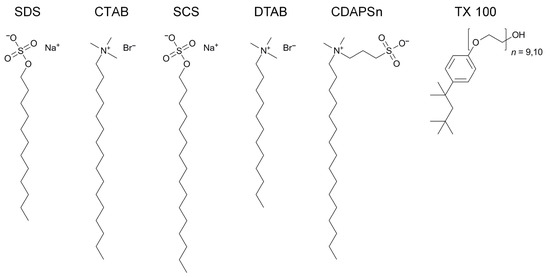
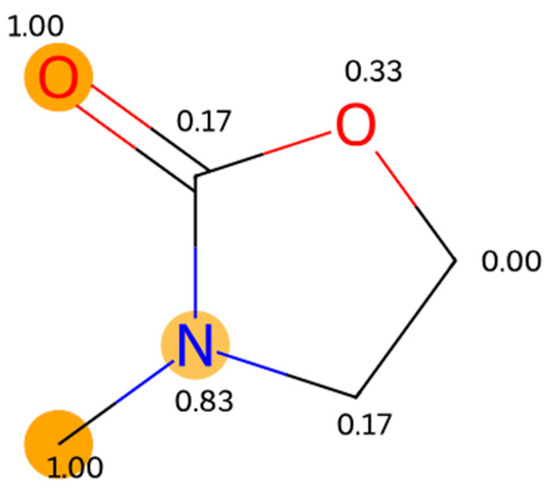
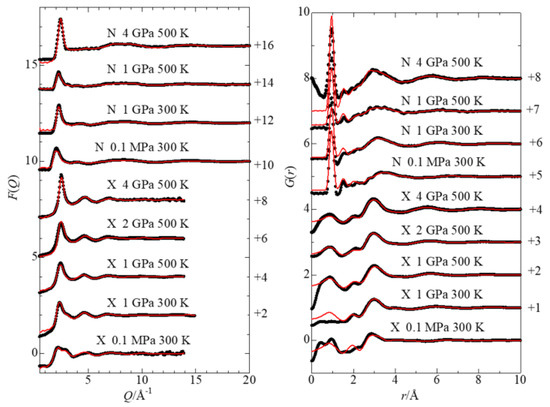
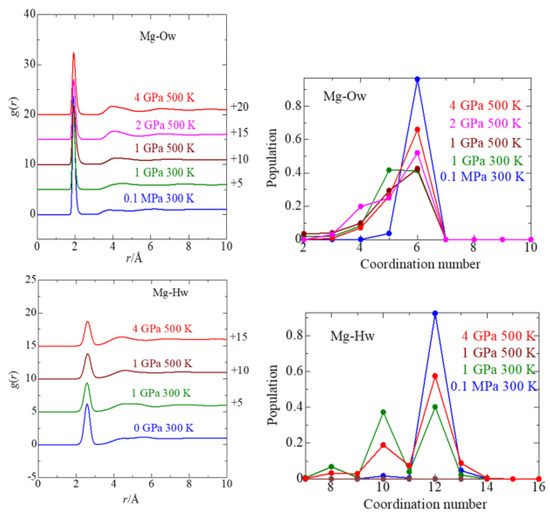
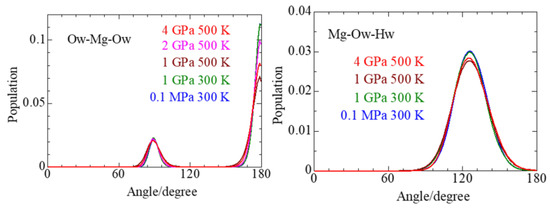
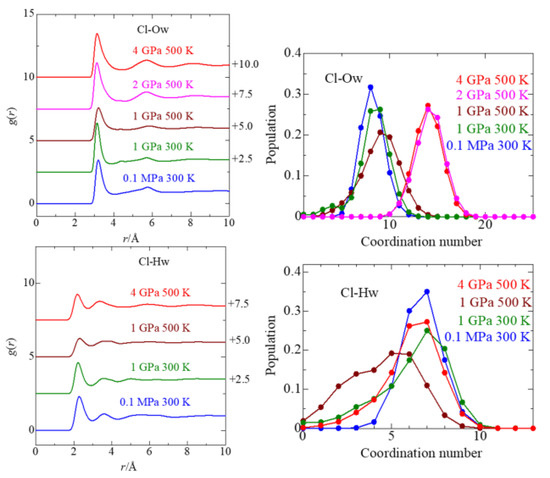
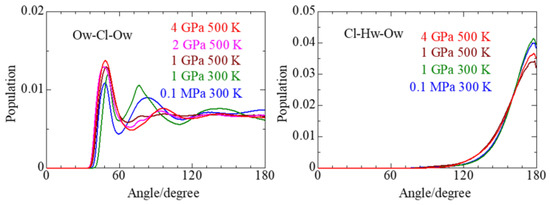
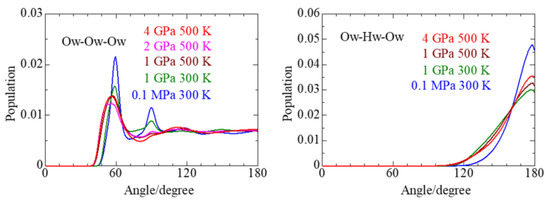
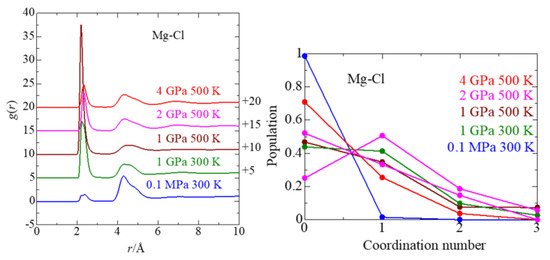
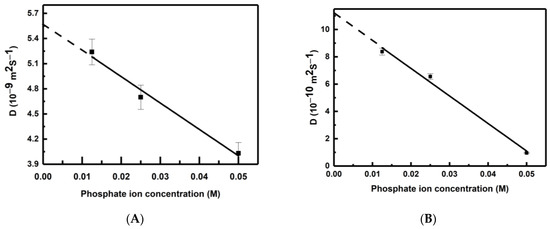
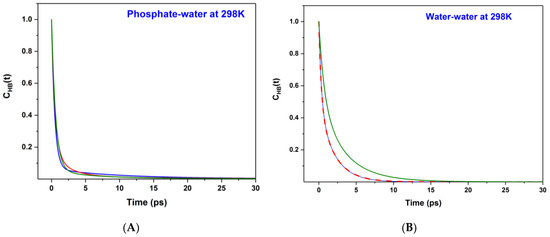
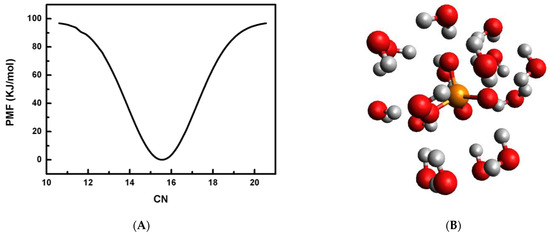
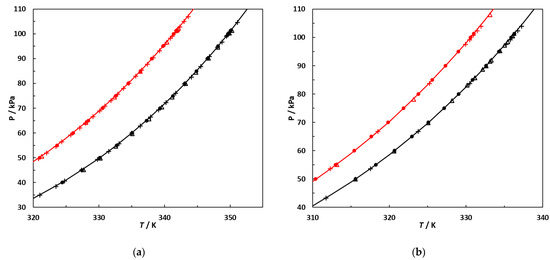
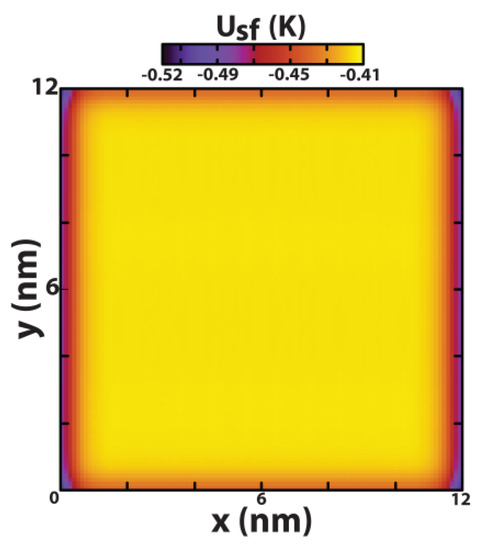
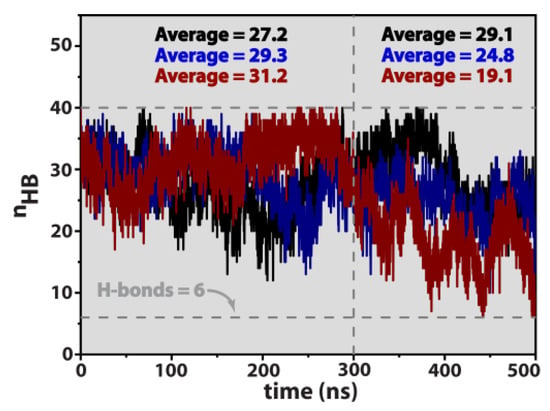
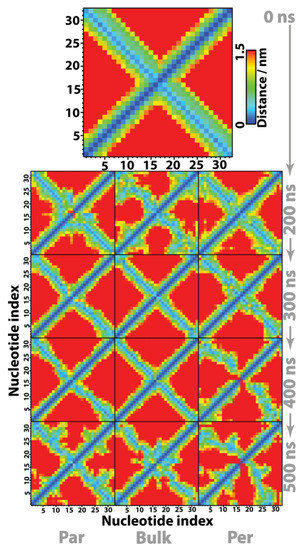
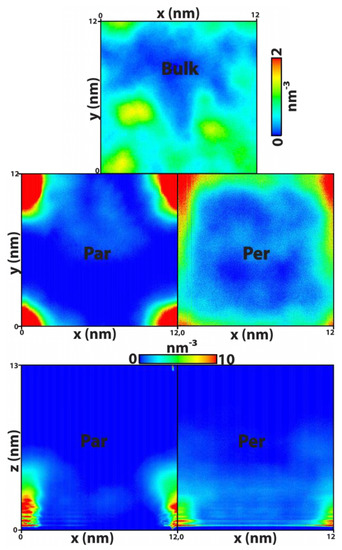
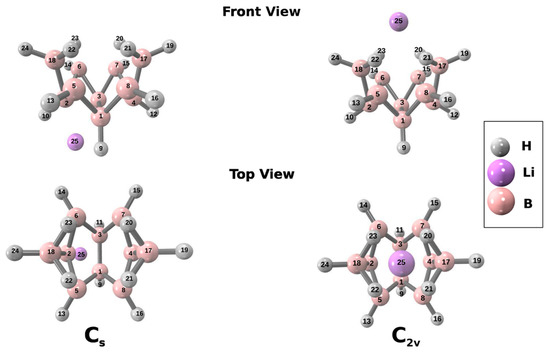
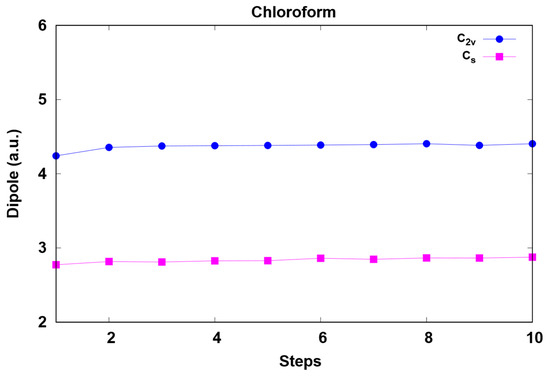
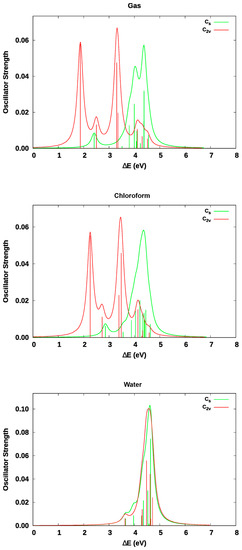
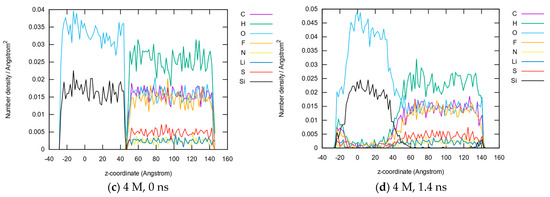
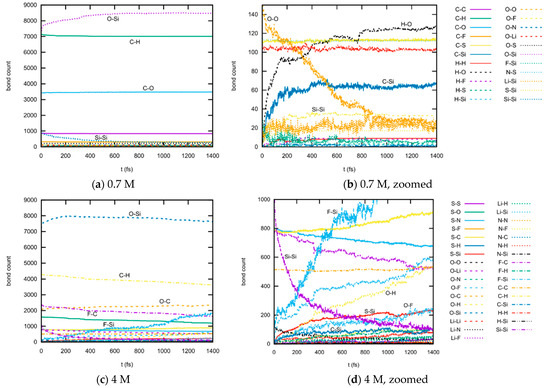
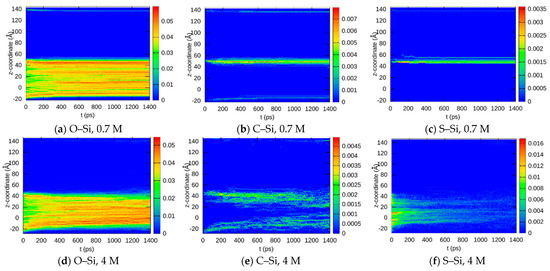
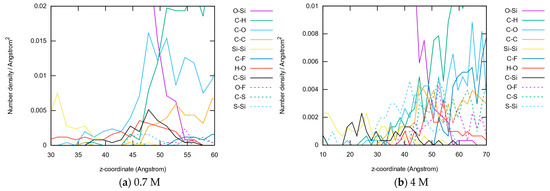
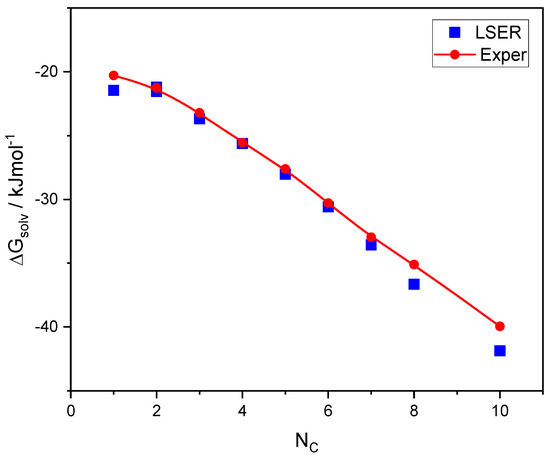
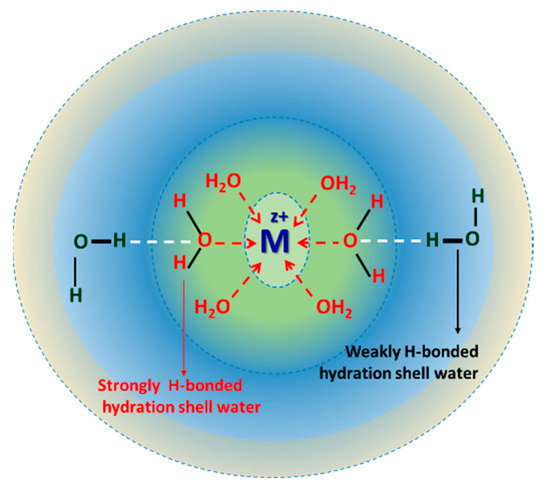
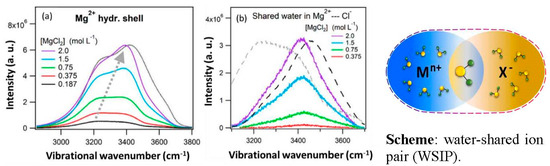
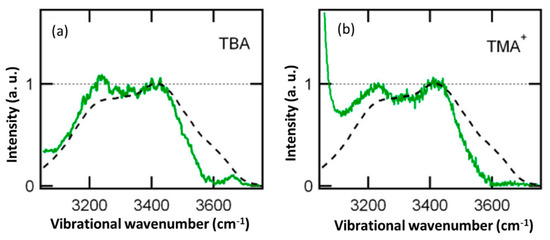
This article summarizes a series of seventeen publications by the authors devoted to molecular dynamics modeling of various indicator dyes (molecular probes) enclosed in surfactant micelles. These dyes serve as generally recognized tools for studying various types of organized solutions, among which surfactant micelles in water are the simplest and most explored. The modeling procedure involves altogether 50 to 95 surfactant molecules, 16 to 28 thousand water molecules, and a single dye molecule. The presentation of the simulation results was preceded by a brief review of the state of experimental studies. This article consists of three parts. First, despite numerous literature data devoted to modeling the micelles itself, we decided to revisit this issue. The structure and hydration of the surface of micelles of surfactants, first of all of sodium
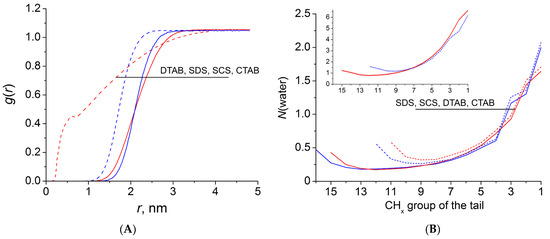
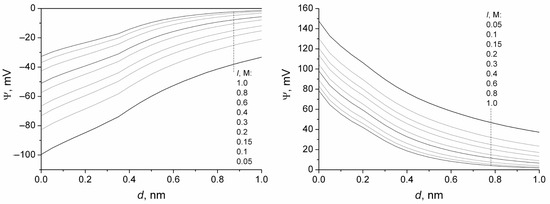
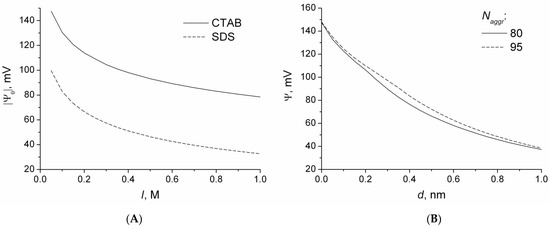
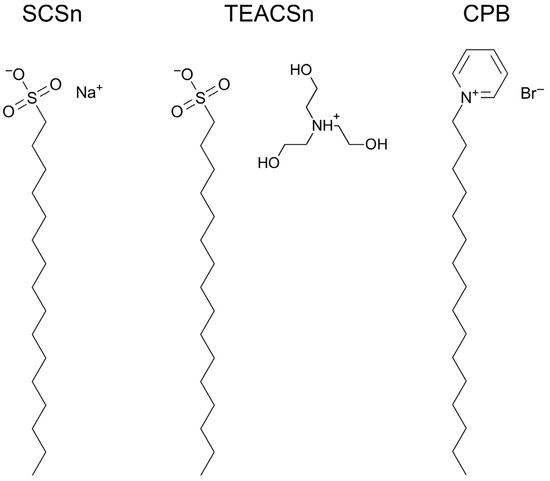
n-dodecylsulfate, SDS, and cetyltrimethylammonium bromide, CTAB, were studied. The values of the electrical potential, Ψ, were estimated as functions of the ionic strength and distance from the surface. The decrease in the Ψ value with distance is gradual. Attempts to consider both DS
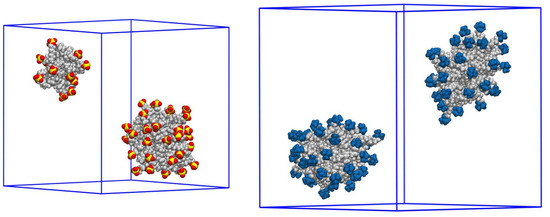
− and CTA
+ micelles in water without counterions result in a decay into two smaller aggregates. Obviously, the hydrophobic interaction (association) of the hydrocarbon tails balances the repulsion of the charged headgroups of these small “bare” micelles. The second part is devoted to the study of seven pyridinium
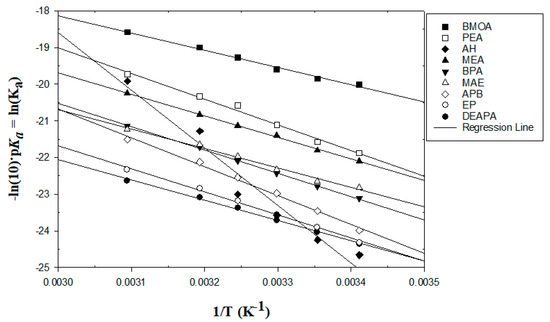
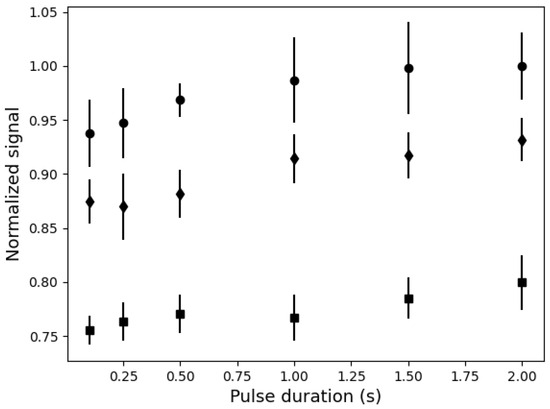
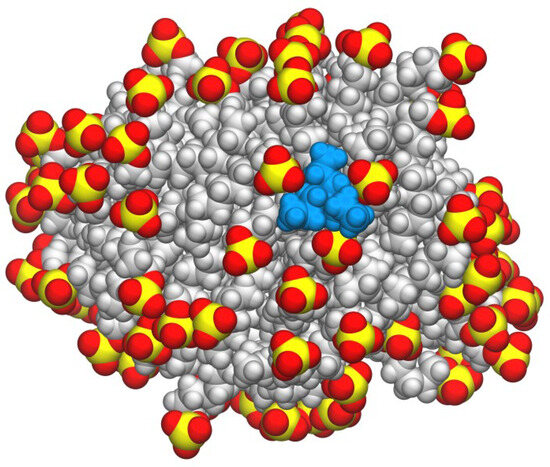
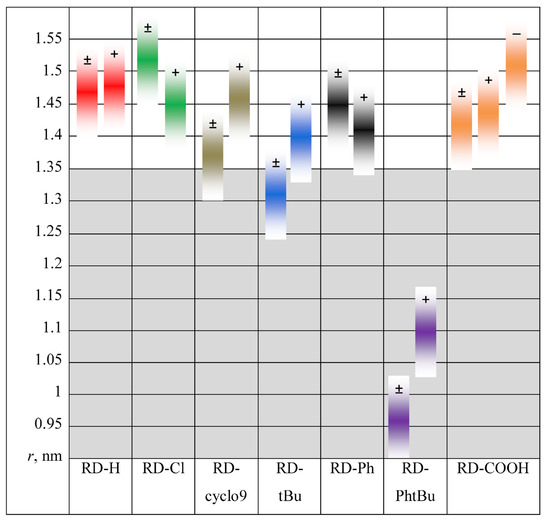
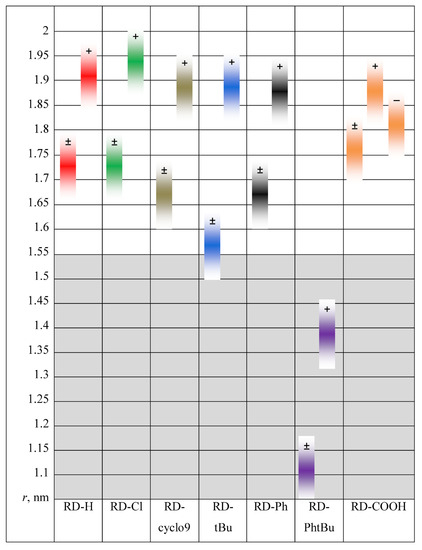
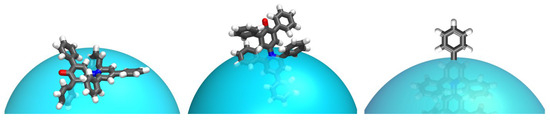
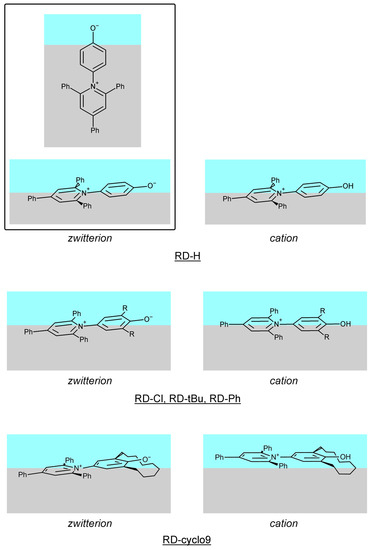
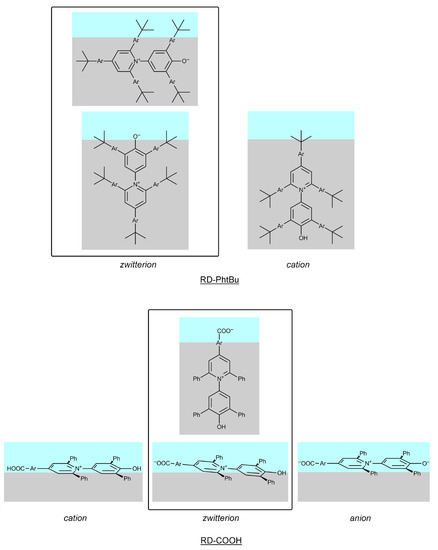
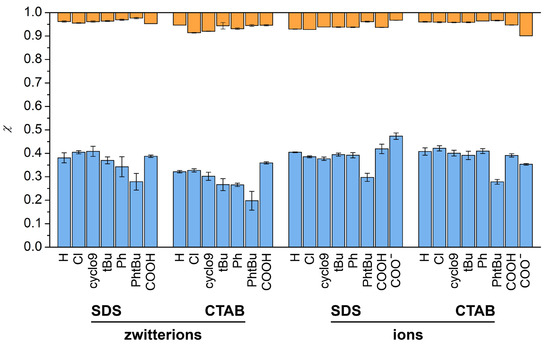
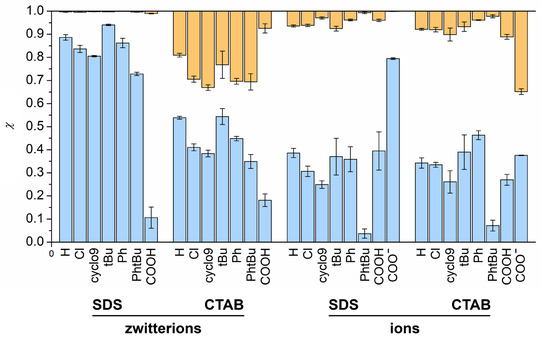
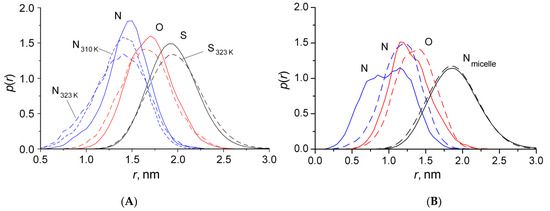
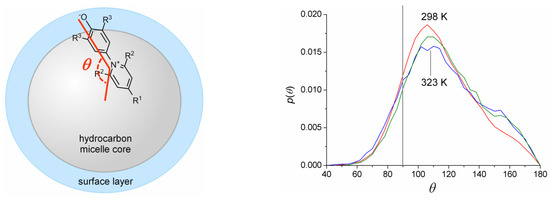
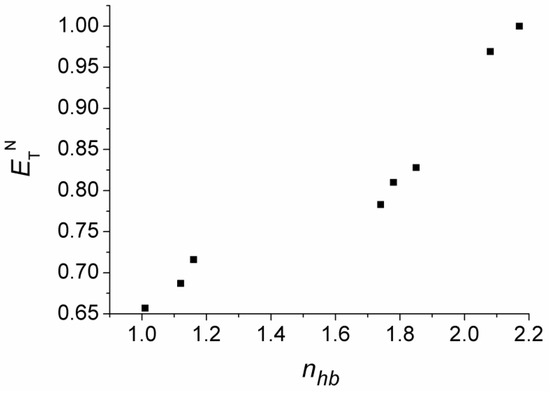
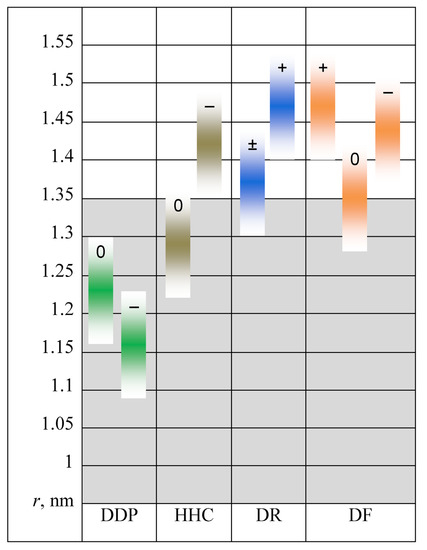
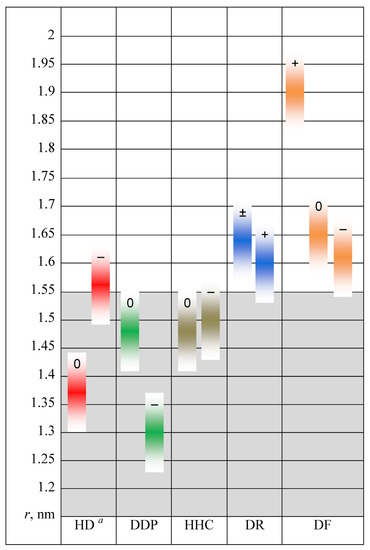
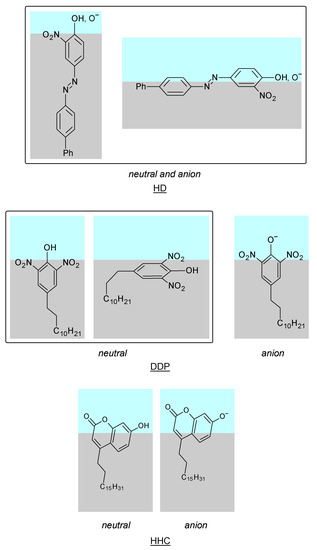
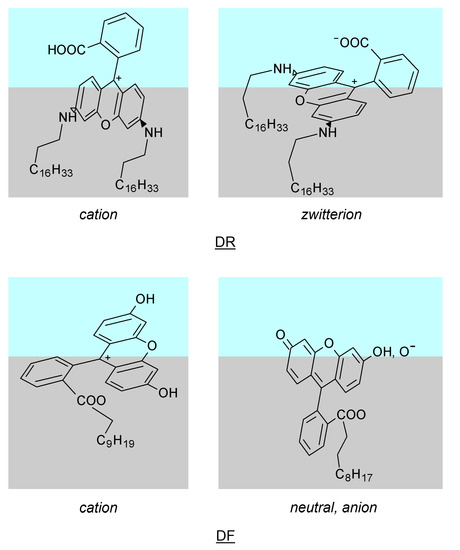
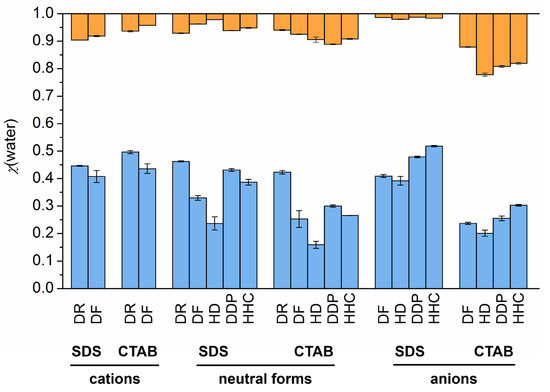
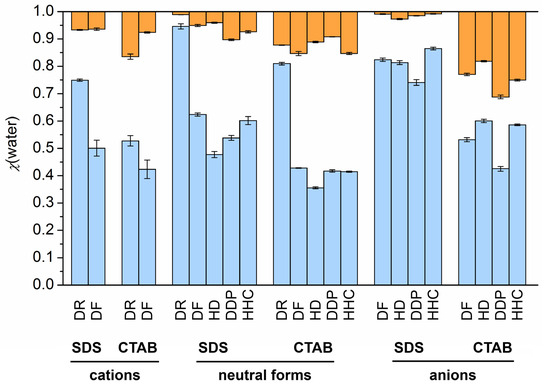
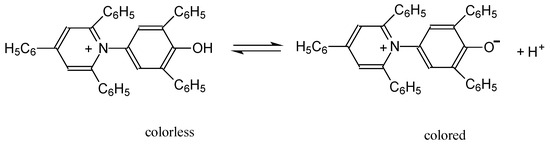
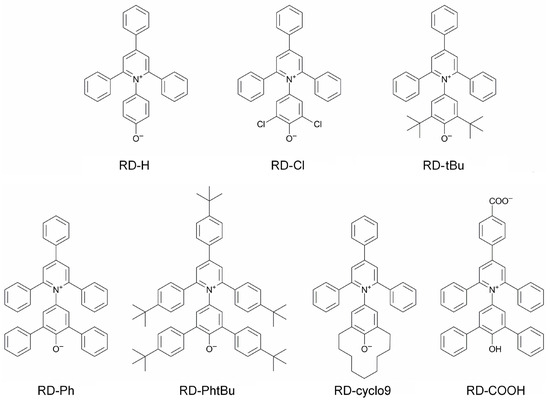
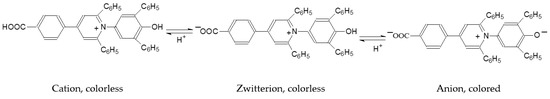
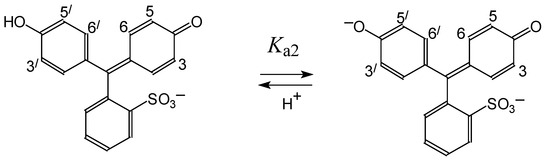
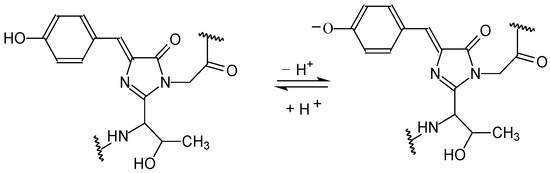
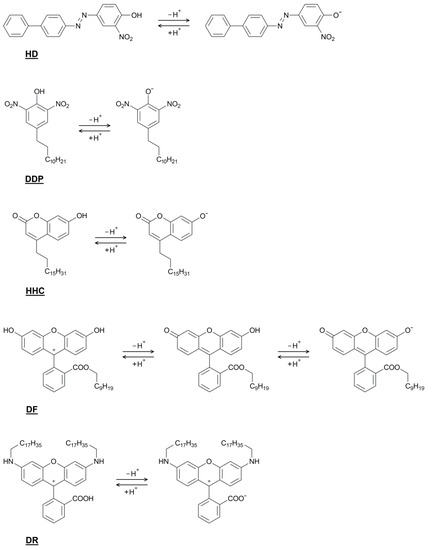
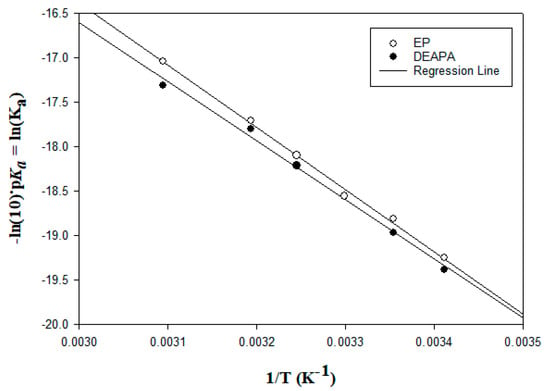
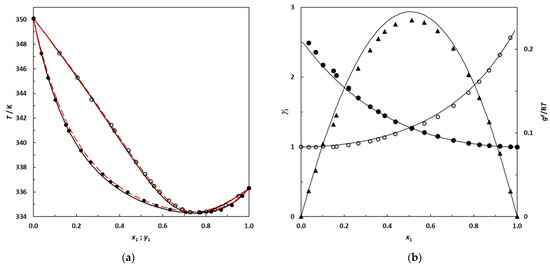
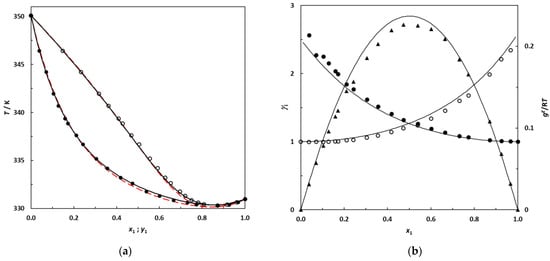
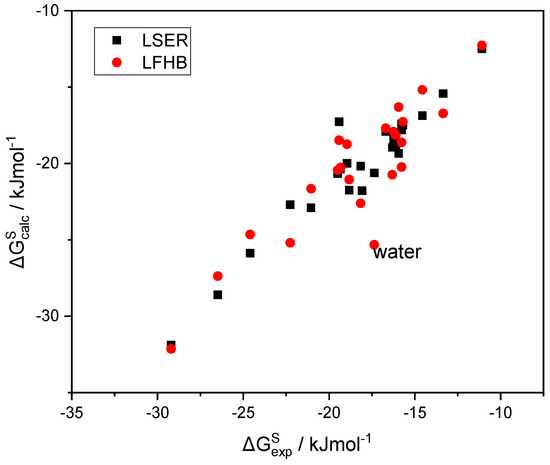
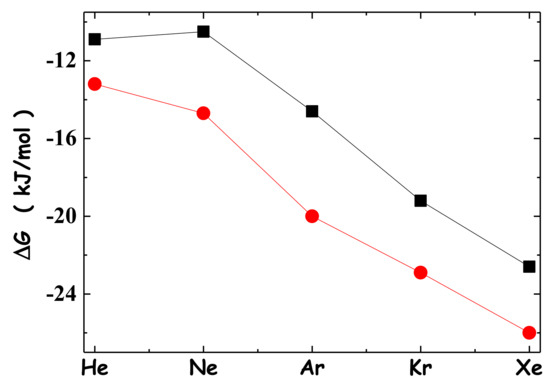
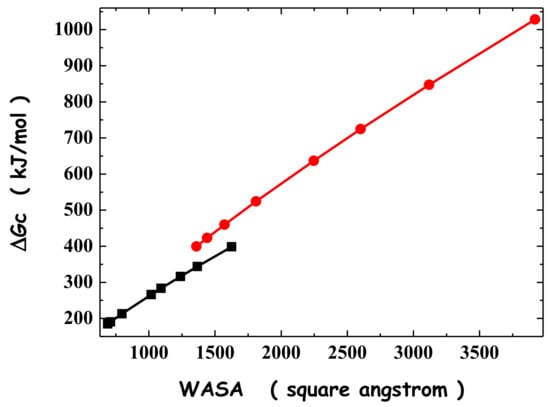
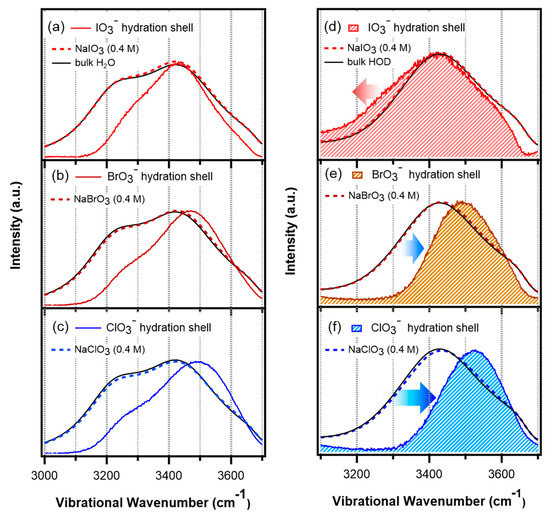
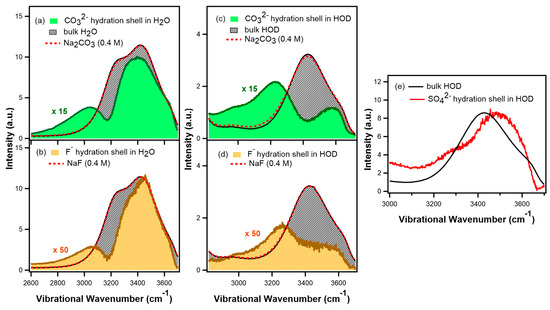
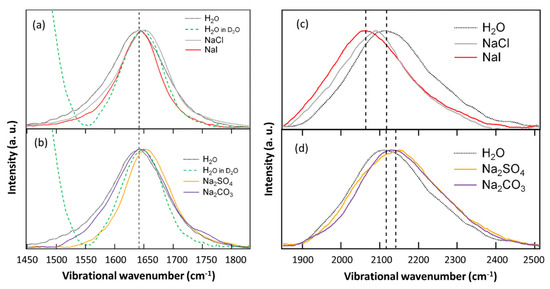
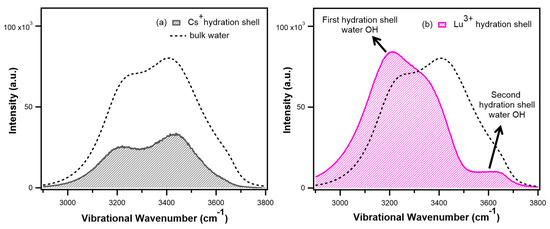
N-phenolates, known as Reichardt’s dyes, in ionic micelles. These most powerful solvatochromic indicators are now used for examining various colloidal systems. The localization and orientation of both zwitterionic and (colorless) cationic forms are generally consistent with intuitive ideas about the hydrophobicity of substituents. Hydration has been quantitatively described for both the dye molecule as a whole and the oxygen atom. A number of markers, including the visible absorption spectra of Reichardt’s dyes, enable assuming a better hydration of the micellar surface of SDS than that of CTAB. However, our data show that it is more correct to speak about the more pronounced hydrogen-bonding ability of water molecules in anionic micelles than about better hydration of the SDS micelles as compared to CTAB ones. Finally, a set of acid–base indicators firmly fixed in the micellar pseudophase were studied by molecular dynamics. They are instruments for estimating electrostatic potentials of micelles and related aggregates as Ψ
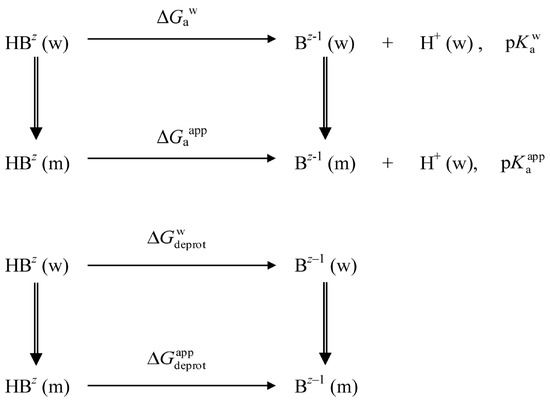
, where
and
are indices of so-called intrinsic and apparent dissociation constants. In this case, in addition to the location, orientation, and hydration, the differences between values of
and indices of the dissociation constants in water were estimated. Only a semi-quantitative agreement with the experimental data was obtained. However, the differences between
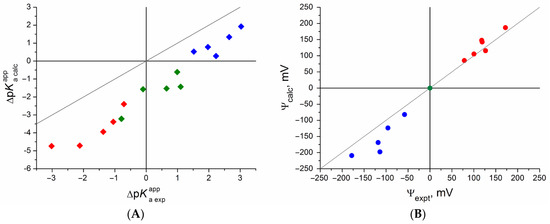
of a given indicator in two micellar solutions do much better agree with the experimental data. Accordingly, the experimental Ψ values of ionic micelles, as determined using the
in nonionic micelles as
, are reproduced with reasonable accuracy for the corresponding indicator. However, following the experimental data, a scatter of the Ψ values obtained with different indicators for given micelles is observed. This problem may be the subject of further research.
Full article











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}