


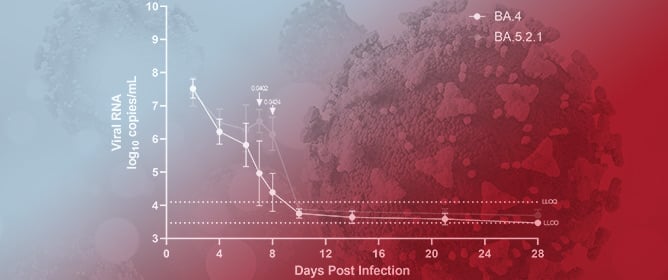
by
, , , , , and


Viruses 2023, 15(9), 1866; https://doi.org/10.3390/v15091866 (registering DOI) - 01 Sep 2023
Abstract
RNA viruses are often cited as a significant factor affecting the populations of both domestic honey bees and wild pollinators. To expedite the development of effective countermeasures against these viruses, a more comprehensive understanding of virus biology necessitates extensive collaboration among scientists from
[...] Read more.
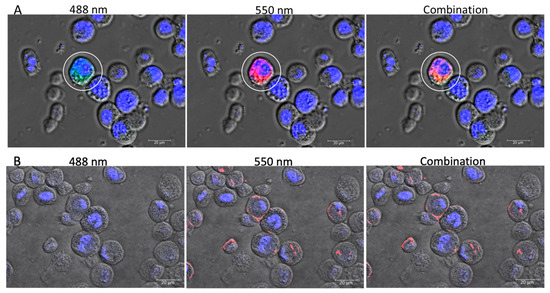
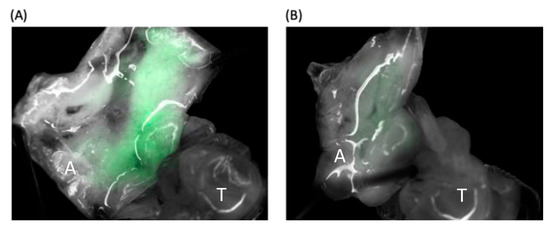
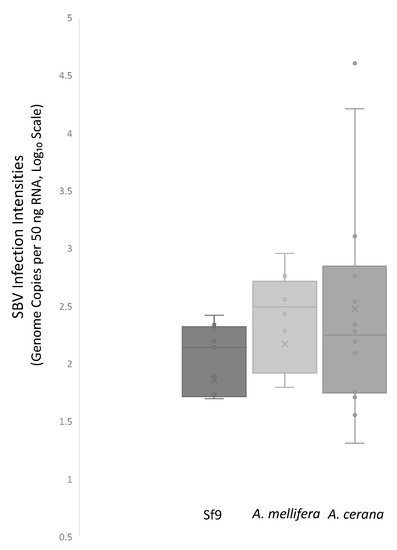
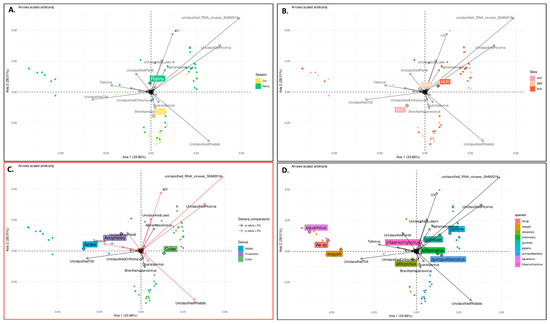
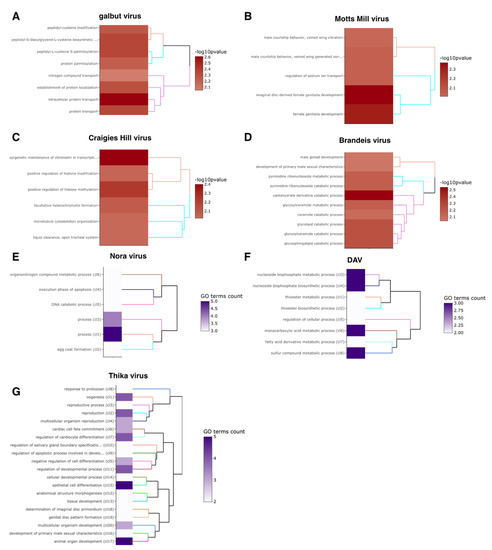
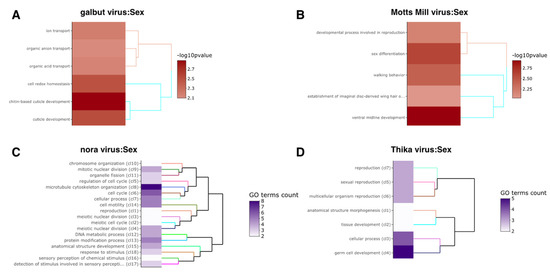
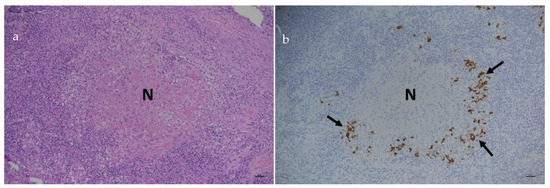
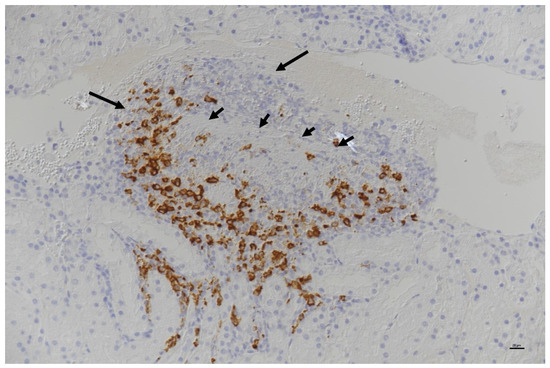
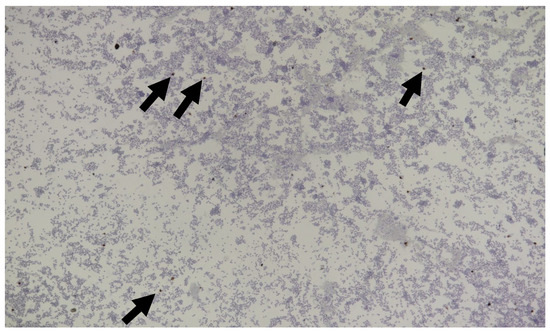
RNA viruses are often cited as a significant factor affecting the populations of both domestic honey bees and wild pollinators. To expedite the development of effective countermeasures against these viruses, a more comprehensive understanding of virus biology necessitates extensive collaboration among scientists from diverse research fields. While the infectious virus clone is a robust tool for studying virus diseases, the current methods for synthesizing infectious clones of bee-infecting RNA viruses entail the in vitro transcription of the viral genome RNA in 8–10 kb, presenting challenges in reproducibility and distribution. This article reports on the synthesis of an infectious clone of the Chinese variant sacbrood virus (SBV) using a DNA plasmid containing an Autographa californica multiple nucleopolyhedrovirus (AcMNPV) immediate-early protein (IE1) promoter to trigger transcription of the downstream viral genome within hosts. The results demonstrate that the IE1-SBV plasmid can synthesize SBV clones in a widely used lepidopteran immortal cell line (Sf9) and honey bee pupae. Furthermore, the negative strand of the clone was detected in both Sf9 cells and honey bee pupae, indicating active infection and replication. However, the transfection of Sf9 cells was observed in only a limited proportion (less than 10%) of the cells, and the infection did not appear to spread to adjacent cells or form infective virions. The injection of honey bee pupae with 2500 ng of the IE1-SBV plasmid resulted in high infection rates in Apis cerana pupae but low rates in A. mellifera pupae, although the dosage was comparatively high compared with other studies using in vitro transcribed viral RNA. Our findings suggest that the synthesis of bee-infecting RNA viruses using DNA plasmids is feasible, albeit requiring additional optimization. However, this method holds substantial potential for facilitating the production of clones with various sequence modifications, enabling the exploration of viral gene functions and biology. The ease of distributing infectious clones in DNA plasmid form may foster collaboration among scientists in applying the clone to bee biology, ecology, and behavior, ultimately offering a comprehensive approach to managing virus diseases in the future.
Full article
(This article belongs to the Section Insect Viruses)
►
Show Figures
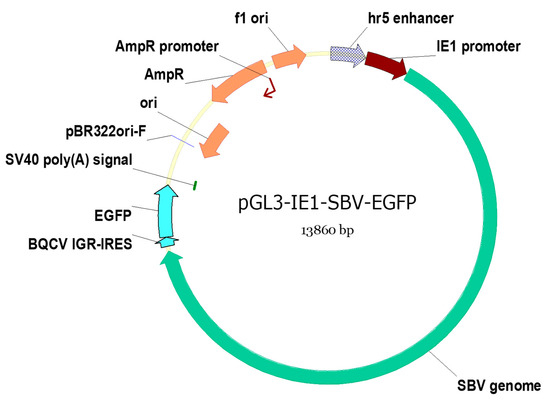
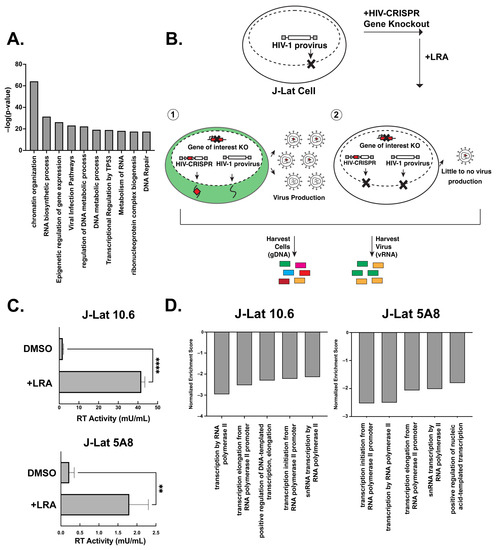
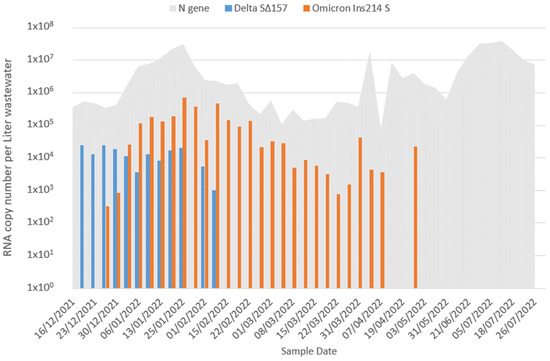
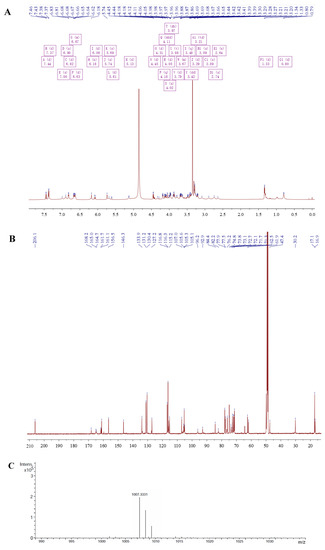
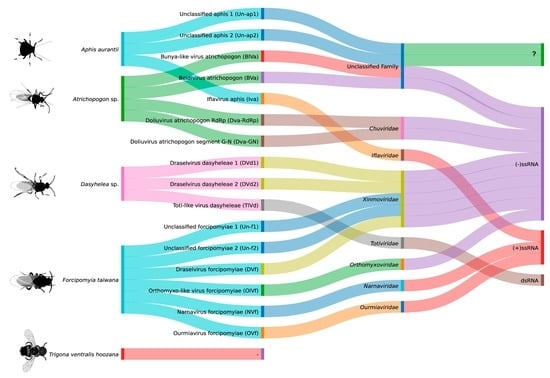
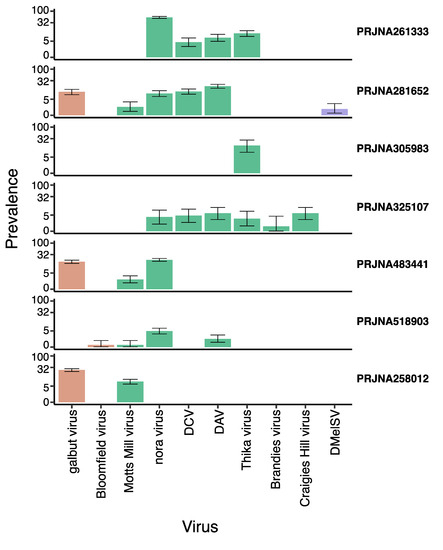
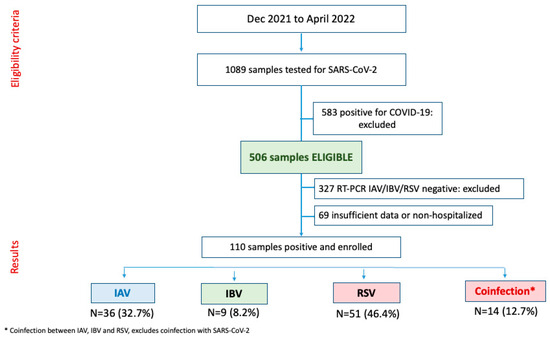
Figure 1

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}